





CopyRight 2009-2020 © All Rights Reserved.版权所有: 中国海关未经授权禁止复制或建立镜像

进口黄水仙基于RAD-Seq的SNP标记开发与分析
作者:于文涛1 张明哲2 沈建国1 袁向芬3 尹文秀2 吕继洲3*
摘 要 为对海量进口黄水仙鳞茎观赏花卉进行快速高精准鉴定,开发其特异性单核苷酸多态性分子标记位点,本研究通过对4种不同进口黄水仙品种和中国水仙共18个样品进行高通量简化基因组测序,得到进口黄水仙简化基因组信息;通过构建简化基因组文库和分子标记检测,得到简化基因组有效数据97.33 G,开发出群体单核苷酸多态性分子标记12532个;通过主成分分析和构建遗传关系邻接树分析,可高分辨率鉴定出所有参试水仙属鳞茎花卉品种。利用高通量简化基因组测序技术,可高效率、低成本地开发大量用于进口黄水仙鳞茎花卉精准鉴定的单核苷酸多态性分子标记,为海关在口岸执法把关提供技术支撑。
于文涛1 张明哲2 沈建国1 袁向芬3 尹文秀2 吕继洲3*
摘 要 为对海量进口黄水仙鳞茎观赏花卉进行快速高精准鉴定,开发其特异性单核苷酸多态性分子标记位点,本研究通过对4种不同进口黄水仙品种和中国水仙共18个样品进行高通量简化基因组测序,得到进口黄水仙简化基因组信息;通过构建简化基因组文库和分子标记检测,得到简化基因组有效数据97.33 G,开发出群体单核苷酸多态性分子标记12532个;通过主成分分析和构建遗传关系邻接树分析,可高分辨率鉴定出所有参试水仙属鳞茎花卉品种。利用高通量简化基因组测序技术,可高效率、低成本地开发大量用于进口黄水仙鳞茎花卉精准鉴定的单核苷酸多态性分子标记,为海关在口岸执法把关提供技术支撑。
关键词 黄水仙;简化基因组测序;单核苷酸多态性;分子鉴定;鳞球茎花卉
Development and Analysis of SNP Loci for Imported Narcissus pseudonarcissus Based on RAD-seq
YU Wen-Tao1 ZHANG Ming-Zhe2 SHEN Jian-Guo1YUAN Xiang-Fen3 YIN Wen-Xiu2 LV Ji-Zhou3*
Abstract In order to accurately identify the imported scale bulbs of Narcissus pseudonarcissus, single nucleotide polymorphism (SNP) mapping sites were developed. This work obtained the SNP loci in Narcissus spp. by restriction-site associated DNA sequencing (RAD-seq) that performed in twelve samples for four cultivars of N. pseudonarcissus and six samples of N. tazetta var. chinensis. By building the genome library and examining of stacks, this study has obtained 97.33 G clean data and 12532 SNP loci. All tested cultivars could be accurately identified by the analysis of principal component and the neighbor-joined tree of genetic relationship. It suggests that the RAD-seq can develop a mass of SNP markers for identification efficiently. Furthermore, this study could offer technical supports for the law enforcement of customs in ports.
Keywords Narcissus pseudonarcissus; RAD-seq; SNP; molecular identification; scale bulb
Abstract In order to accurately identify the imported scale bulbs of Narcissus pseudonarcissus, single nucleotide polymorphism (SNP) mapping sites were developed. This work obtained the SNP loci in Narcissus spp. by restriction-site associated DNA sequencing (RAD-seq) that performed in twelve samples for four cultivars of N. pseudonarcissus and six samples of N. tazetta var. chinensis. By building the genome library and examining of stacks, this study has obtained 97.33 G clean data and 12532 SNP loci. All tested cultivars could be accurately identified by the analysis of principal component and the neighbor-joined tree of genetic relationship. It suggests that the RAD-seq can develop a mass of SNP markers for identification efficiently. Furthermore, this study could offer technical supports for the law enforcement of customs in ports.
黄水仙(Narcissus pseudonarcissus L.)原产欧洲,为石蒜科(Amaryllidaceae)水仙属(Narcissus L.)著名观赏花卉[1]。为满足人民群众对观赏花卉的多样性需求,我国大量进口黄水仙鳞茎。同时,为解决我国水仙品种单一、种性退化等问题,从国外引进优良水仙花种质资源已成为当前较为有效的方法之一[2]。从2016年开始,我国进口黄水仙等鳞球茎花卉的数量超过5亿株,价值超过1亿美元,且进口鳞球茎花卉的数量和贸易额呈增长态势[3]。对进口黄水仙等鳞球茎花卉的具体物种和品种信息进行科学准确鉴定,是进一步深入开展其他工作的基础与前提。针对水仙属植物Narcissus spp.(主要包括水仙 N. tazetta var. chinensis M. Roem.和黄水仙 N. pseudonarcissus L.)等观赏花卉,由于其进口时多处于休眠的鳞茎状态,通过肉眼观察和常规显微镜观察等形态学方法难以开展鉴定工作。而之前的分子标记研究主要为AFLP等方法[4-6],操作相对烦琐,一般针对单个种类,数据分析时间较长,难以大范围推广应用。
简化基因组测序技术(Restriction-site associated DNA sequencing, RAD-Seq),是随着二代测序的诞生而发展起来的一种在全基因组酶切的基础上相对简化的全基因组测序技术,其优点在于不依赖参考基因组的信息,一次测序即可获得数以万计的多态性遗传标记,已广泛应用于基因组学、遗传学、生态学、分类学等研究领域。单核苷酸多态性(Single Nucleotide Polymorphism, SNP),是生命个体基因组中丰富且稳定的可遗传多态性,由单个碱基的颠倒或转换引起,也可由单个碱基的插入或缺失所致,具有二态性[7]。以高通量简化基因组测序技术作为开发单核苷酸多态性分子标记的基础,可快速开发相关鉴定分子标记。同时,由于简化基因组测序和单核苷酸多态性技术的高通量、多目标性、富于基因代表性、高遗传稳定性、易实现分析自动化等优点[8-10],能够开展海量进口黄水仙鳞茎花卉的多目标高准确性高通量鉴定,为海关在口岸执法把关提供技术支撑,并进一步达到保护和促进我国花卉产业发展的目的。
1 材料与方法
1.1 材料
实验材料为4个品种的进口黄水仙,分别为Narcissus pseudonarcissus L. 'Tahiti' (ta)、N. pseudonarcissus L. 'Dutch Master' (dm)、N. pseudonarcissus L. 'Ice Follies' (if)和N. pseudonarcissus L. 'Pink Charm' (pc),以中国漳州水仙N. tazetta L. var. chinensis Roem (chinensis)为对照种。其中,4个品种的进口黄水仙于2015~2017年自荷兰引进;各品种的实验材料均选取周径为15~20 cm、花球饱满紧实、形美端正、无病虫害、无损伤破裂、鳞茎外皮光滑的健康鳞茎。进口黄水仙每个品种选取3个实验个体,中国水仙选取6个实验个体,共计18个实验样品。
1.2 方法
1.2.1 DNA提取
采用植物全基因组DNA提取试剂盒对样品进行全基因组DNA提取。
1.2.2 简化基因组测序
将样品全基因组DNA进行酶切,经桥式PCR制备Cluster,随后在HiSeq测序仪上进行测序操作[11]。
1.2.3 构建测序文库
对测序reads结果使用Stampy软件[12]比对,对结果进行进一步mate-paire修复、添加read组信息、对重复reads进行标记等后续处理,随后构建完整的测序文库。
1.2.4 测序评估
对得到的原始数据(Raw data)进行barcode拆分,之后进一步过滤获得有效数据(Clean data)。
1.2.5 单核苷酸多态性检测
采用Stacks 1.4.4[13]对有效数据进行个体read聚类和单核苷酸多态性检测。对所有样本的所有碱基信息进行比对,在一致性序列的基础上,将检测到的基因型与参考序列之间存在多态性的位点进行过滤,得到高可信度的单核苷酸多态性数据集。
1.2.6 基因分型
基于各个样品鉴定得到的高质量单核苷酸多态性位点及其相关基因型,整合该群体中所有相关样品的单核苷酸多态性位点集,并对候选位点进行过滤,主要考虑因素为所有样品中该位点检测的缺失情况,以及所有样品中该位点基因型的多态性,最终生成基因分型列表。
1.2.7 鉴定数据分析
实验使用EIGENSOFT[14]进行主成分分析(PCA),以确认所有样品之间的遗传种群结构和关系,同时用样本和单核苷酸多态性基因座构建的二维矩阵数据进行主成分分析,并计算几个主要特征向量,以及定位这些特征向量中的每个样本。随后,使用TreeBest软件[15]中的邻接算法,经过100次迭代绘制系统邻接树。
2 实验结果
表1 18个水仙属植物样品测序质量
Table 1 Sequencing quality of 18 Nacissus spp. samples
样本名称 | /M | /G | /M | /G | /% | 碱基占比/% | 碱基占比/% |
dm_1 | 37.54 | 5.48 | 16.26 | 2.37 | 45.96 | 97.61 | 93.59 |
dm_2 | 41.79 | 6.1 | 17.57 | 2.56 | 45.96 | 97.55 | 93.44 |
dm_3 | 21.74 | 3.17 | 10.21 | 1.49 | 45.77 | 97.66 | 93.7 |
if_1 | 62.2 | 9.17 | 36.1 | 5.32 | 45.56 | 97.57 | 93.51 |
if_2 | 66.25 | 9.74 | 38.86 | 5.71 | 45.4 | 97.48 | 93.31 |
if_3 | 105.59 | 15.52 | 61.24 | 9 | 45.73 | 97.5 | 93.37 |
pc_1 | 61.51 | 9.04 | 35.88 | 5.27 | 45.79 | 97.5 | 93.37 |
pc_2 | 109.74 | 16.13 | 64.55 | 9.49 | 45.79 | 97.48 | 93.33 |
pc_3 | 69.61 | 10.2 | 39.62 | 5.8 | 45.91 | 97.33 | 92.94 |
ta_1 | 56.43 | 8.32 | 33.2 | 4.9 | 45.83 | 97.51 | 93.38 |
ta_2 | 70.65 | 10.42 | 40.17 | 5.93 | 45.78 | 97.54 | 93.46 |
ta_3 | 116.17 | 17.13 | 66.78 | 9.85 | 46.14 | 97.54 | 93.45 |
chinensis_1 | 36.45 | 5.32 | 17.53 | 2.56 | 47.63 | 97.6 | 93.55 |
chinensis_2 | 90.79 | 13.3 | 49.79 | 7.29 | 47.05 | 97.52 | 93.37 |
chinensis_3 | 52.16 | 7.62 | 27.41 | 4 | 46.9 | 97.57 | 93.49 |
chinensis_4 | 90.85 | 13.31 | 48.78 | 7.15 | 46.86 | 97.49 | 93.31 |
chinensis_5 | 66.98 | 9.81 | 37.99 | 5.57 | 46.28 | 97.51 | 93.38 |
chinensis_6 | 31.63 | 4.62 | 16.18 | 2.36 | 47.29 | 97.54 | 93.42 |
2.1 测序数据统计
18个样品通过简化基因组测序技术构建测序文库,下机原始数据174.4 G,按照barcode拆分、过滤获得有效数据97.33 G。有效数据中进口黄水仙Tahiti_3产出量最高,为9.85G。GC占比在各样品测序结果中均处于正常水平,所有样品中质量≥30碱基占比最低为进口黄水仙Pink Charm_2,其值为92.94%,其他样品均>93%(见表1),为质量较高水平,可用于后续进一步数据分析。
2.2 分子标记开发
质量较高的简化基因组测序结果为单核苷酸多态性分子标记的开发提供了基础和保证,实验共获得12532个单核苷酸多态性位点用于高级分析(见表2)。在18个样品中,4个品种的进口黄水仙样品的单个样品基因分型数量均远超中国水仙的单个样品基因分型数量。进口黄水仙样品中基因分型总数最多的为N. pseudonarcissus L. 'Pink Charm'。同一品种内不同个体间的差异较小。
表2 18个水仙属植物样品SNP信息
Table 2 The SNP information statistics of 18 Nacissus spp. samples
样品名称 | 总数 | 总数 | 总数 | 纯合占比/% | /% |
dm_1 | 7008 | 5303 | 1705 | 75.67 | 24.33 |
dm_2 | 7116 | 5254 | 1862 | 73.83 | 26.17 |
dm_3 | 5505 | 4453 | 1052 | 80.89 | 19.11 |
if_1 | 8716 | 5929 | 2787 | 68.02 | 31.98 |
if_2 | 9066 | 5960 | 3106 | 65.74 | 34.26 |
if_3 | 9253 | 5848 | 3405 | 63.2 | 36.8 |
pc_1 | 8817 | 5849 | 2968 | 66.34 | 33.66 |
pc_2 | 9363 | 5632 | 3731 | 60.15 | 39.85 |
pc_3 | 8934 | 5820 | 3114 | 65.14 | 34.86 |
ta_1 | 7909 | 5781 | 2128 | 73.09 | 26.91 |
ta_2 | 8428 | 5950 | 2478 | 70.6 | 29.4 |
ta_3 | 9003 | 5967 | 3036 | 66.28 | 33.72 |
chinensis_1 | 740 | 663 | 77 | 89.59 | 10.41 |
chinensis_2 | 758 | 674 | 84 | 88.92 | 11.08 |
chinensis_3 | 751 | 662 | 89 | 88.15 | 11.85 |
chinensis_4 | 508 | 463 | 45 | 91.14 | 8.86 |
chinensis_5 | 673 | 594 | 79 | 88.26 | 11.74 |
chinensis_6 | 524 | 474 | 50 | 90.46 | 9.54 |
2.3 主成分分析及邻接树鉴定率
主成分分析是研究群体结构和进行鉴定的重要方法,是使用正交变换将可能相关变量的一组观察值转换成被称为主成分的线性不相关变量值的统计过程。主成分的数量小于或等于原始变量的数量。在实验结果的主成分分析图(见图1)中,所有进口黄水仙品种样品均位于X轴原点左侧,而中国水仙品种样品均位于X轴原点右侧,两个物种差异较大,可以完全区分鉴别。同时,进口黄水仙4个不同品种内的3个个体也各自聚类,且与其他品种无交集。经过主成分分析,所有样品的鉴定率均为100%,效果较好。
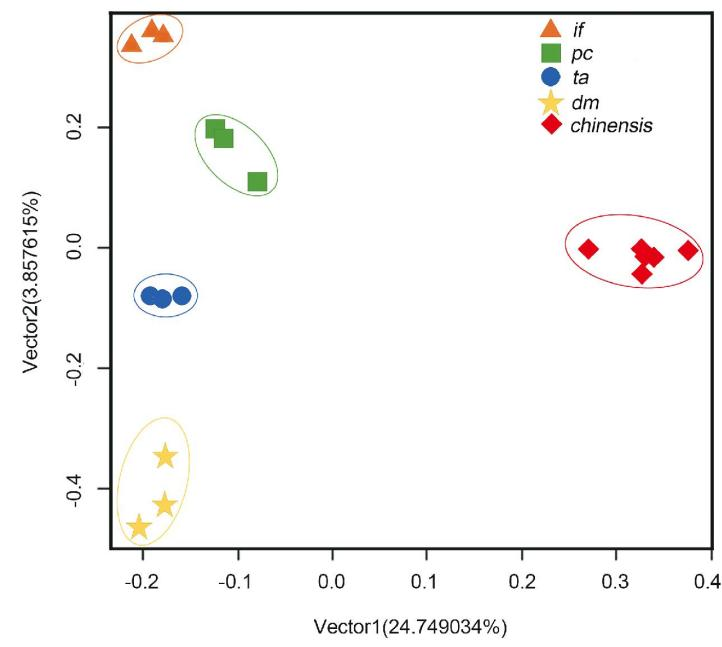
图1 18个水仙属植物样品PCA分析图
Fig.1 Principal component analysis for 18 Nacissus spp. Samples
系统树是生物信息学中描述不同生物之间相互关系的方法,也是检测分子标记鉴定率的重要方法[16]。系统由结点和分支组成,每一结点表示一个分类学单元(属、种群、个体等),系统分支定义了分类单元之间的关系。本研究构建的邻接树(见图2)共分为两大支,其中,所有的进口黄水仙样品聚为一支,中国水仙的6个样品聚为另一支。进口黄水仙样品的一支又分为2个分支,其中,N. pseudonarcissus L. 'Dutch Master'的3个样品单独聚为一支,其他3个品种聚为另一支,且这3个品种在分支中由各自的3个样品分别聚为单系分支。本研究中各个品种的所有样品均单独聚为一支,且均为单系群,因此,本研究开发的单核苷酸多态性分子标记系统树法的鉴定率为100%,鉴定准确率高。
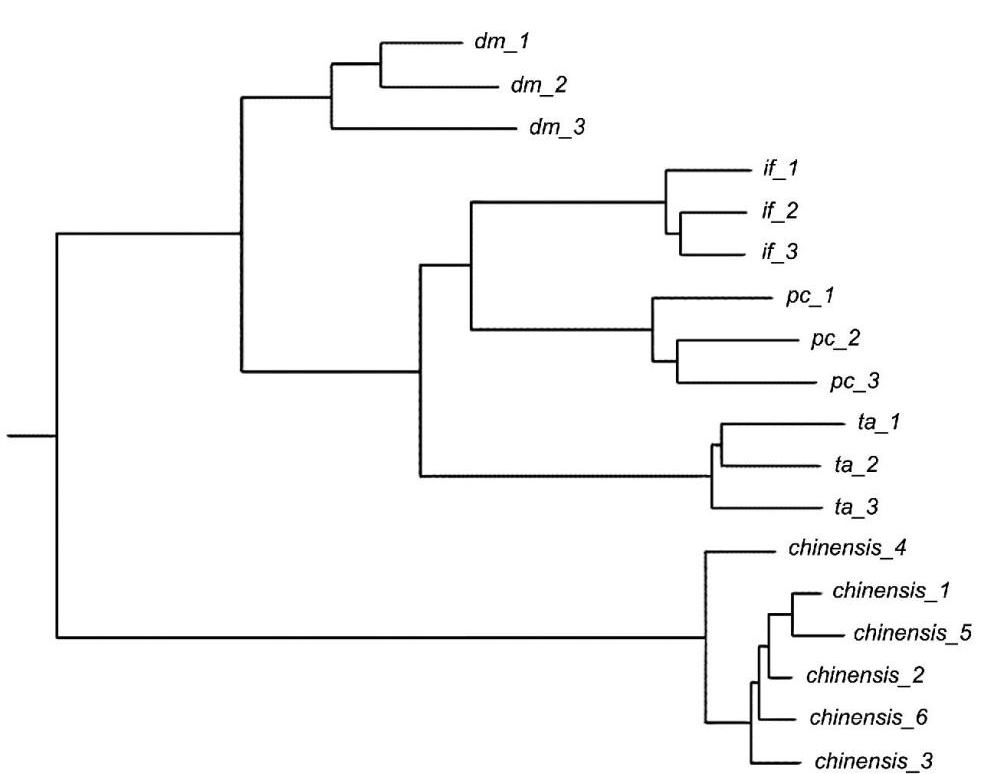
图2 18个水仙属植物样品NJ系统树图
Fig.2 Dendrogram of 18 Nacissus spp. Samples
3 讨论
近年来,高通量测序技术的进步带动了分子生物学在基因组领域研究的全面发展[17-19],而且为单核苷酸多态性等分子标记提供了高效率开发的基础[20-21]。其中,基于高通量简化基因组测序技术开发特异性高的单核苷酸多态性分子标记已在大量物种中应用[22-23],可以获得高密度的单核苷酸多态性分子标记,并具有较高的有效性和一致性。此前,针对水仙属植物研究的分子标记主要为AFLP、RAPD等[4, 24],然而这些方法获得分子标记的效率很低,且使用过程中操作烦琐,自动化程度较低。单核苷酸多态性作为最新一代分子标记,可实现自动化分析,检测效率高[10, 21, 25]。
目前,已发表论文暂未有水仙属植物单核苷酸多态性开发的相关报告。本研究首次将简化基因组测序技术应用于水仙属植物的单核苷酸多态性分子标记的开发,得到有效数据97.33 G,有效reads 658 M,测序数据的平均Q30为93.41%,平均GC含量为46.20%,整体测序质量较高。利用这些测序数据,研究团队开发出群体单核苷酸多态性分子标记12532个,其中进口黄水仙开发的单核苷酸多态性分子标记数量远超于中国水仙,且二者的遗传距离较远,说明这两个同属物种的遗传进化或选育体系存在较大差异。在本研究中,91.6%的进口黄水仙样品单核苷酸多态性分子标记的杂合率高于20%,58.3%的进口黄水仙样品单核苷酸多态性分子标记的杂合率高于30%,而中国水仙的所有样品杂合率均低于12%,说明进口黄水仙品种具有高度杂合性,相对而言,中国水仙的杂合性较低。
通过主成分分析与邻接树法两种方法,研究团队对获得的单核苷酸多态性分子标记的鉴定效率进行了验证。在两种方法中,所有样品的鉴定准确率均为100%。这些分子标记不但可以将进口黄水仙与中国水仙两个物种进行区分,而且可以精确区分鉴定所有参试进口黄水仙的品种,说明所开发的单核苷酸多态性分子标记用于进口黄水仙鳞茎鉴定是完全可行的。下一步,研究团队将对得到的分子标记进行筛选,并确认针对特定品种的特异性分子标记,以期进一步提高鉴定效率。
4 结论
本研究通过简化基因组测序技术,以4个进口黄水仙品种和中国水仙的鳞茎为实验对象,进行单核苷酸多态性分子标记位点开发。通过全基因组DNA提取、二代测序、测序文库构建、单核苷酸多态性检测和基因分型等步骤,获得97.33 G高质量的有效数据和12532个单核苷酸多态性位点。另外,我们通过主成分分析和邻接树构建两种方法,以获得的单核苷酸多态性位点对实验样品进行鉴定,可以精准鉴定所有参试样品的品种。由此可见,将简化基因组测序技术开发用于进口黄水仙鳞茎精准鉴定的单核苷酸多态性分子标记是高效可行的。
【该文经CNKI学术不端文献检测系统检测,总文字复制比为5.6%】。
参考文献
[1] TERRY M I, RUIZ-HERNáNDEZ V, ÁGUILA D J, et al. The Effect of Post-harvest Conditions in Narcissus sp. Cut Flowers Scent Profile [J]. Frontiers in plant science, 2021, 11: 2144.
[2]卞阿娜, 潘东明. 洋水仙在漳州地区的引种筛选研究 [J]. 热带作物学报, 2013, 34(8): 1444-1449.
[3]董燕. 2019年我国花卉进出口数据统计分析[J].中国花卉园艺, 2020, 11: 24-25.
[4] TUCCI G, WINFIELD M, D‘AMATO G, et al. Genetic diversity in Narcissus poëticus L. and N. radiiflorus Salisb.(Amaryllidaceae) in two different populations: AFLP and karyological studies [J]. Caryologia, 2004, 57(4): 405-411.
[5] LIU C, ZHANG X. AFLP and SCAR Markers Analysis on the Dual Color Flower Mutants of Gladiolus [J]. Acta Agriculturae Boreali-Occidentalis Sinica, 2009, 5.
[6] NOY-PORAT T, FLAISHMAN M, ESHEL A, et al. Florogenesis of the Mediterranean geophyte Narcissus tazetta and temperature requirements for flower initiation and differentiation [J]. Scientia horticulturae, 2009, 120(1): 138-142.
[7] LAFRAMBOISE T. Single nucleotide polymorphism arrays: a decade of biological, computational and technological advances [J]. Nucleic acids research, 2009, 37(13): 4181-4193.
[8] UENO S, UCHIYAMA K, MORIGUCHI Y, et al. Scanning RNA-Seq and RAD-Seq approach to develop SNP markers closely linked to MALE STERILITY 1 (MS1) in Cryptomeria japonica D. Don [J]. Breeding science, 2019, 69(1): 19-29.
[9] DíAZ-ARCE N, RODRíGUEZ-EZPELETA N. Selecting RAD-Seq data analysis parameters for population genetics: the more the better? [J]. Frontiers in genetics, 2019, 10: 533.
[10] ANDREWS K R, ADAMS J R, CASSIRER E F, et al. A bioinformatic pipeline for identifying informative SNP panels for parentage assignment from RAD seq data [J]. Molecular ecology resources, 2018, 18(6): 1263-1281.
[11] CAPORASO J G, LAUBER C L, WALTERS W A, et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms [J]. The ISME journal, 2012, 6(8): 1621-1624.
[12] LUNTER G, GOODSON M. Stampy: a statistical algorithm for sensitive and fast mapping of Illumina sequence reads [J]. Genome research, 2011, 21(6): 936-939.
[13] CATCHEN J, HOHENLOHE P A, BASSHAM S, et al. Stacks: an analysis tool set for population genomics [J]. Molecular ecology, 2013, 22(11): 3124-3140.
[14] SHRINER D. Investigating population stratification and admixture using eigenanalysis of dense genotypes [J]. Heredity, 2011, 107(5): 413-420.
[15] VILELLA A J, SEVERIN J, URETA-VIDAL A, et al. EnsemblCompara GeneTrees: Complete, duplication-aware phylogenetic trees in vertebrates [J]. Genome research, 2009, 19(2): 327-335.
[16] BESTAGINI P, TAGLIASACCHI M, TUBARO S. Image phylogeny tree reconstruction based on region selection; proceedings of the 2016 IEEE International Conference on Acoustics, Speech and Signal Processing (ICASSP), F, 2016 [C]. IEEE.
[17] ANDERS S, PYL P T, HUBER W. HTSeq—a Python framework to work with high-throughput sequencing data [J]. Bioinformatics, 2015, 31(2): 166-169.
[18] QI Y, ZANG S-Q, WEI J, et al. High-throughput sequencing provides insights into oral microbiota dysbiosis in association with inflammatory bowel disease [J]. Genomics, 2021, 113(1): 664-676.
[19] REUTER J A, SPACEK D V, SNYDER M P. High-throughput sequencing technologies [J]. Molecular cell, 2015, 58(4): 586-597.
[20] ZHU Z, SUN B, LEI J. Specific-Locus Amplified Fragment Sequencing (SLAF-Seq) as High-Throughput SNP Genotyping Methods [M]. Crop Breeding. Springer. 2021: 75-87.
[21] ARCA M, MARY-HUARD T, GOUESNARD B, et al. Deciphering the genetic diversity of landraces with high-throughput SNP genotyping of DNA bulks: methodology and application to the maize 50k array [J]. Frontiers in plant science, 2021, 11: 1926.
[22]ULASZEWSKI B, MEGER J, BURCZYK J. Comparative Analysis of SNP Discovery and Genotyping in Fagus sylvatica L. and Quercus robur L. Using RADseq, GBS, and ddRAD Methods [J]. Forests, 2021, 12(2): 222.
[23] STOJANOVA B, ŠURINOVá M, ZEISEK V, et al. Low genetic differentiation despite high fragmentation in the endemic serpentinophyte Minuartia smejkalii (M. verna agg., Caryophyllaceae) revealed by RADSeq SNP markers [J]. Conservation Genetics, 2020, 21(2): 187-198.
[24] LU G, ZHANG X, ZOU Y, et al. Effect of radiation on regeneration of Chinese narcissus and analysis of genetic variation with AFLP and RAPD markers [J]. Plant cell, tissue and organ culture, 2007, 88(3): 319-327.
[25] SYVäNEN A-C. Toward genome-wide SNP genotyping [J]. Nature genetics, 2005, 37(6): S5-S10.
(文章类别:CPST-C)
