





CopyRight 2009-2020 © All Rights Reserved.版权所有: 中国海关未经授权禁止复制或建立镜像

分散固相萃取-UPLC-MS/MS法测定动物肝脏中左旋咪唑的残留量
作者:李晓岩1 巩志国1 房 芳1 巴哈提古丽middot;马那提拜1 孙 鲁1
摘 要 研究了采用1%冰乙酸-乙腈溶液分散固相萃取( QuEChERS),建立了超高效液相色谱-串联质谱法测定动物肝脏中左旋咪唑残留量的分析方法。样品提取后,用无水硫酸钠(Na2SO4)及氨基(NH2)吸附剂净化,多反应监测扫描模式(MRM)分析,采用外标法定量。实验结果表明,左旋咪唑在10~200 μg/L线性范围内具有良好的线性关系,相关系数(r2)为:0.9996。测定低限(LOQ)为10 μg/kg。试验以空白样品为基体做加标回收,测得左旋咪唑的回收率在71.3%~105.5%,相对标准偏差(n=6)为5.2%~8.1%。满足动物肝脏中左旋咪唑残留量的快速分析要求。
李晓岩1 巩志国1 房 芳1 巴哈提古丽·马那提拜1 孙 鲁1
摘 要 研究了采用1%冰乙酸-乙腈溶液分散固相萃取( QuEChERS),建立了超高效液相色谱-串联质谱法测定动物肝脏中左旋咪唑残留量的分析方法。样品提取后,用无水硫酸钠(Na2SO4)及氨基(NH2)吸附剂净化,多反应监测扫描模式(MRM)分析,采用外标法定量。实验结果表明,左旋咪唑在10~200 μg/L线性范围内具有良好的线性关系,相关系数(r2)为:0.9996。测定低限(LOQ)为10 μg/kg。试验以空白样品为基体做加标回收,测得左旋咪唑的回收率在71.3%~105.5%,相对标准偏差(n=6)为5.2%~8.1%。满足动物肝脏中左旋咪唑残留量的快速分析要求。
关键词 固相萃取;UPLC-MS/MS;左旋咪唑;动物肝脏
Determination of Levamisole Residues in Animal Liver by Dispersive Solid Phase Extraction-UPLC-MS/MS
LI Xiao-Yan1 GONG Zhi-Guo1 FANG-Fang1 BAkhetgu Manatbay1 SUN Lu1
Abstract The study was established an analytical method for the determination of levamisole residues in animal liver by dispersive solid phase extraction-ultra high performance liquid chromatography-tandem mass spectrometry. The samples were extracted with 1% glacial acetic acid-acetonitrile solution, purified with anhydrous sodium sulfate (Na2SO4) and amino (NH2) adsorbents, analyzed by multi-reaction monitoring scanning mode (MRM), and quantified by external standard method. Levamisole had a good linear relationship in the linear range of 10-200 μg/L, and the correlation coefficient (r2) was 0.9996. The lower limit of determination (LOQ) was 10 μg/kg. In the test, the blank sample was used as the matrix to do standard recovery. The recovery rate of levamisole was 71.3%-105.5%, and the relative standard deviation (n=6) was 5.2%-8.1%. Meet the requirements of rapid analysis of levamisole residues in animal livers.
Keywords solid phase extraction; UPLC-MS/MS; levamisole; animal liver
左旋咪唑是一种人工合成的广谱驱虫药,应用历史较长。主要起到驱除猪、牛、羊等胃肠道中的线虫和肺丝虫[1-3],并有效预防和治疗鸡球虫病及绵羊脑包虫等疾病,在养殖业中广泛应用。但是左旋咪唑具有潜在的基因毒性,使用该药后对动物的消化系统、神经系统、心血管系统及造血系统、泌尿系统和皮肤都有不同程度的毒副作用,如果长期、大量使用或盲目用药将会导致动物源性食品中残留大量左旋咪唑,人食用后残留药物在人体内长时间累积,会造成人体多种器官和系统产生严重不良反应。为保证动物源性食品的安全性,加拿大、美国、日本和欧盟等许多发达国家都严格制定了的限量标准10~100 μg/kg[4],我国也在动物源性食品兽药最大残留限量中规定,牛、猪、羊等动物源性食品中左旋咪唑的最大残留限量(MRL)为10~100 μg/kg。并将左旋咪唑药物列入了《中华人民共和国动物及动物源性食品残留物质监控计划》中。所以建立一种高效、简便、准确、低成本的检测方法对动物源性食品安全有着极其重要的意义。我国检测左旋咪唑残留量的主要方法有比旋光度法、高效液相色谱法(HPLC)、紫外分光光度法[5-9]、一阶导数光谱法、气相色谱法[10-11](GC)、液相色谱串联质谱法[12](LC-MS/MS)、GB 29681-2013、NY/T 3139-2017、GB/T 22994-2008。目前,国内研究左旋咪唑治疗方面与药理作用的报道较多,但有关左旋咪唑在动物肝脏中残留量的检测方法未见报道,也无相关的检测标准。本文采用了分散固相萃取 ( QuEChERS) 样品前处理法,用UPLC-MS/MS检测动物肝脏中左旋咪唑的残留量具有检出限低、灵敏度高、分析结果准确等优点,并为修制定动物源性食品中左旋咪唑检测标准提供技术支撑。
1 实验与方法
1.1 仪器和试剂
超高效液相色谱-质谱联用仪(ACQUITY UPLC-Quattro premierXETM,美国Waters公司);漩涡混合器(VS-2500M,无锡沃信仪器制造有限公司);腕式振荡器(美国BURRELL公司);氮吹仪(美国Organomation 公司);高速离心机(JIDI-20D,广州吉迪仪器有限公司);0.2 μm再生纤维素滤膜(美国Waters公司)。
左旋咪唑标准物质(100 mg,美国Dr. Ehrenstorfer GmbH公司);乙腈和甲醇为色谱纯,均购自Fisher 公司;无水硫酸钠和冰乙酸为分析纯,购自广州化学试剂工厂;氨基(NH2)吸附剂,实验用水均为高纯水,样品来源为实验室日常检验样品。
左旋咪唑标准储备溶液(100 mg/L):精密称取左旋咪唑标准物质0.025g于250 mL容量瓶中,用甲醇溶液定容至刻度。
左旋咪唑标准工作溶液:用甲醇+水(2∶8,v/v)溶液将上述标准储备溶液逐级稀释成适当浓度的标准工作液。
1.2 仪器条件及实验方法
1.2.1 色谱条件
色谱柱:Kromasil Eternity-2.5-C18(2.1 mm×100 mm,5 μm,美国Waters公司);柱温35℃,样品室温度6℃;流量0.25 mL/min,进样量5 μL;流动相:0.1%(体积分数)甲酸为A溶液,甲醇为B溶液;采用梯度洗脱程序:0~3.5 min,A由90%~75%,B由10%~25%;3.5~5.5 min时间段 A由75%~90%保持1 min。运行时间为6.5 min。
1.2.2 质谱条件
采用多反应监测扫描模式(MRM),电喷雾离子源ESI(+),离子源温度:110℃;锥孔反吹气流量:48 L/Hr;毛细管电压:3.5 kV;脱溶剂气流量:550 L/Hr;脱溶剂气温度为350 ℃,驻留时间20 ms;氩气为碰撞气。其他质谱分析参数见表1。
表1 多反应监测扫描模式质谱参数
Table 1 Multi-reaction monitoring scan mode mass spectrometry parameters
化合物 | m/z | m/z | U/V | E/eV | |
左旋咪唑 | 205 | 122.9 | 35 | 29 | |
178.1* | 21 | ||||
注:*为定量离子 | |||||
1.2.3 样品提取
取可食部分动物肝脏(羊肝、牛肝、猪肝及鸡肝)作为样品,经捣碎机充分捣碎,混匀混合研磨仪加工成浆状备用,处理后的样品置于-18 ℃冰箱冷冻保存。
称取5.00 g试样,置于50 mL离心管中,加入15 mL 1%冰乙酸-乙腈溶液,置于腕式振荡器中振荡20 min,然后加入6 g 无水硫酸钠,用涡旋仪高速涡旋混匀1 min,再用高速离心机在8000 r/min条件下离心5 min,备用。
1.2.4 样品净化
称取600 mg NH2吸附剂至另一个新的50 mL离心管中,将1.2.4中的上清液转移至50 mL离心管中,涡旋仪快速涡旋1 min,用高速离心机8000 r/min离心5 min后吸取3 mL上清液于5 mL玻璃试管中,用氮吹仪45℃下氮气缓缓吹干后用1 mL甲醇+水(2+8)溶解残渣,用0.2 μm有机滤膜进行过滤并置于2 mL的样品瓶中,待测。
2 结果与讨论
2.1 样品前处理条件的优化
动物肝脏含有丰富的蛋白质、脂肪和碳水化合物等,并且组成成分比较复杂。乙腈溶液能够使动物肝脏中蛋白质变性并产生沉淀,再用高速离心机离心去除变性后的蛋白质。无水硫酸钠能有效吸收动物肝脏基质中的水分,采用NH2吸附剂吸附动物肝脏基质中部分脂肪和碳水化合物等。在本次的试验中对提取液进行了选择,试验发现,采用乙腈溶液作为提取液,左旋咪唑提取效率差,乙腈和冰乙酸混合溶液明显能提高左旋咪唑提取效率。因此,选择试验乙腈-冰乙酸溶液浓度分别为0.2%、0.5%、1%的进行。实验结果表明,乙腈-冰乙酸(1%)作为提取液可以有效提高左旋咪唑的萃取效率,同时,左旋咪唑峰形都能够达到最佳,所以在本试验中选用浓度为1%的冰乙酸-乙腈作为提取液。
2.2 色谱条件的优化
2.2.1 色谱柱的优化
分别选择了Kromasil Eternity-2.5-C18(2.1 mm× 100 mm)和ACQUITY UPLC BEH HILIC 1.7 μm(2.1 mm×150 mm)两种色谱柱进行实验。结果表明,Kromasil Eternity-2.5-C18色谱柱对左旋咪唑能完全分离,同时,色谱峰型对称,无杂峰干扰。左旋咪唑的保留时间为2.8 min。标准溶液谱图见图1。
2.2.2 流动相的选择
本文主要考察了有机相和水相混合的流动相,其中有机相选择了甲醇溶液和乙腈溶液,水相选择了冰乙酸溶液、乙酸铵溶液和0.1%甲酸水溶液。经多次排列组合试验发现,当使用甲醇溶液和0.1%甲酸水溶液作为流动相时,待测物的峰形尖锐对称,灵敏度较高。因此本文选用流动相0.1%甲酸水溶液为A,甲醇溶液为B。
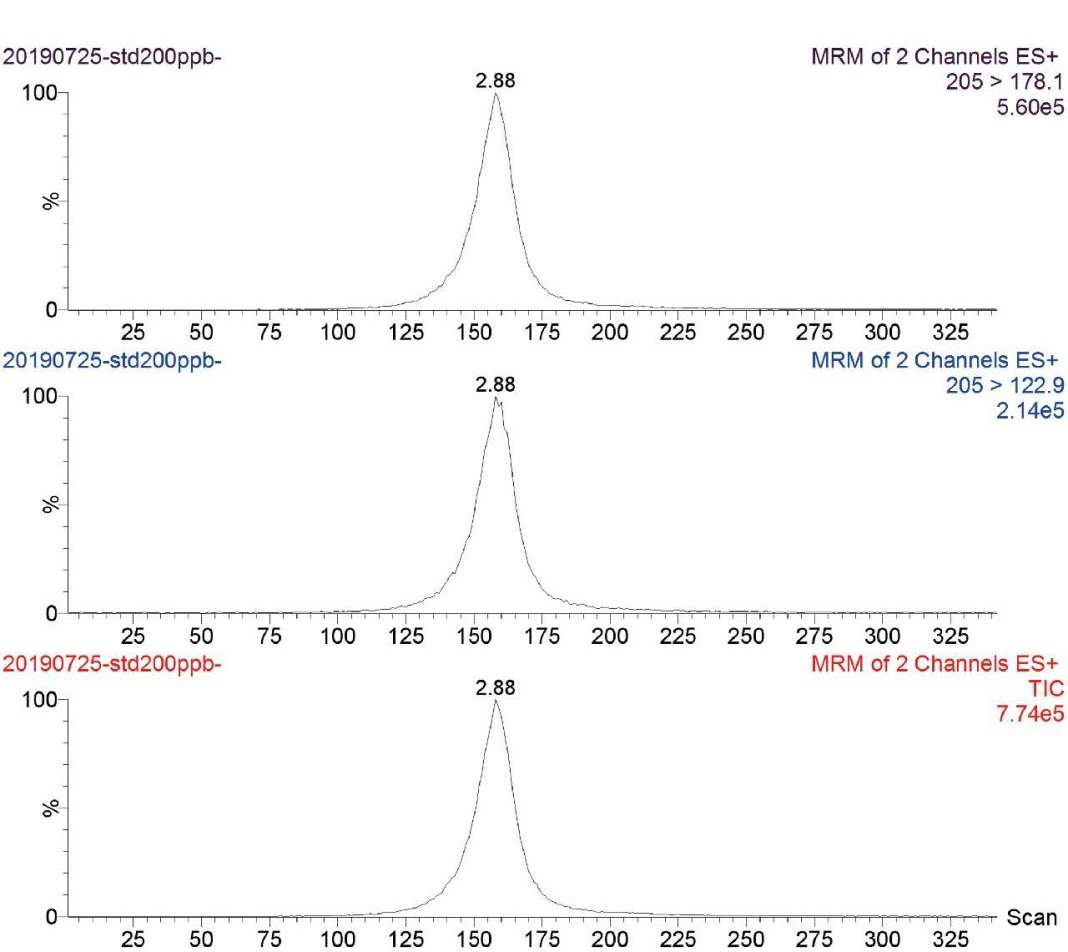
图1 左旋咪唑标准溶液谱图
Fig.1 Levamisole standard solution spectrum
2.3 质谱条件的优化
通过ESI(+)正离子模式进行一级质谱全扫描,对0.7 mg/L左旋咪唑标准溶液进行扫描,得到左旋咪唑的母离子205(m/z),然后再次全扫描左旋咪唑子离子,选择多反应监测(MRM)扫描模式进行分析。同时对锥孔电压和碰撞能量进行调谐及优化,使母离子和子离子的丰度比达到最佳状态,响应信号值达到最强,最终获得左旋咪唑的质谱检测参数(见表1)。
2.4 线性关系及检出限
按照1.1中左旋咪唑标准工作溶液配制方法,配制系列左旋咪唑标准工作溶液,其质量浓度分别为10.0、20.0、50.0、100.0、200.0 μg/L,并依次上机测定,可以得到左旋咪唑的线性关系及相关系数(见表2)。
表2 左旋咪唑的线性方程及相关系数
Table 2 linear equation and correlation coefficient of Levamisole
化合物 | 线性范围 (μg/L) | 线性方程 | 相关系数 (r2) |
左旋咪唑 | 10.0~200.0 | Y=1822.03X+1147.48 | 0.9996 |
当特征离子色谱峰的信号响应强度与基线噪声之比(S/N)≥3时可得到方法检出限,方法定量限为当特征离子色谱峰信号的响应强度与基线噪声之比(S/N)≥10时,可得到方法定量限。经计算分别得到左旋咪唑的方法检出限为3.0 μg/kg,方法定量限为10.0 μg/kg。
2.5 加标回收率及精密度
取猪肝、羊肝、牛肝、鸡肝4种实际样品(实测值为未检出),做3个水平的添加回收试验,3个水平梯度的浓度分别为10.0 μg/kg、20.0 μg/kg、50.0 μg/kg,每组试验重复6次 (n=6),测定回收率及相对标准偏差结果见表3。图2为空白猪肝样品添加10.0 μg/kg后的选择离子流色谱图。
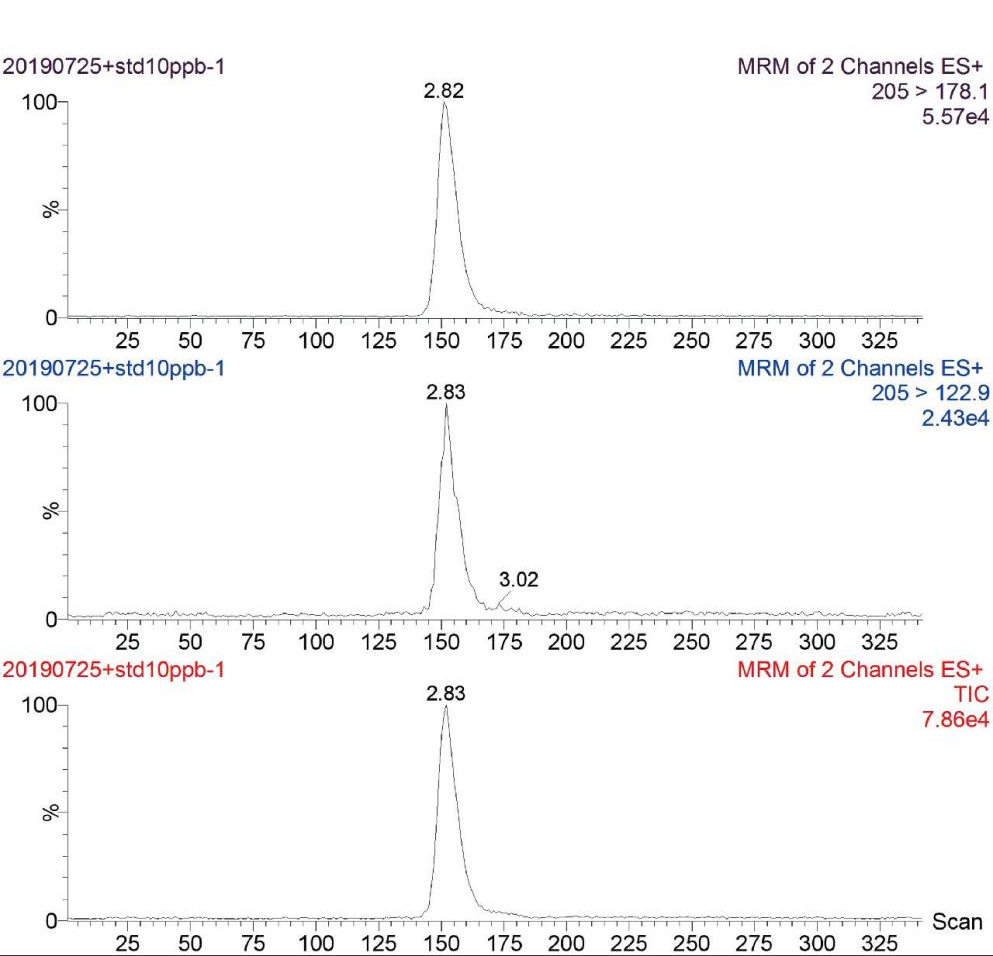
图2 空白猪肝样品添加10.0 μg/kg后的选择离子流色谱图
Fig.2 Selected ion chromatogram after adding 10.0 μg/kg of blank pig liver sample
表3 左旋咪唑在4种动物肝脏中的加标回收率及相对标准偏差RSD(n=6)
Table 3 Standard recovery rate and relative standard deviation of levamisole in four animal livers (n=6)
样 品 | 添加水平 | 回收率范围 | 相对标准偏差 |
猪肝 | 10 | 77.6~88.3 | 5.8 |
20 | 86.6~95.5 | 7.5 | |
50 | 81.2~100.1 | 8.1 | |
羊肝 | 10 | 85.4~96.1 | 4.2 |
20 | 71.3~92.5 | 7.3 | |
50 | 85.2~105.1 | 6.0 | |
牛肝 | 10 | 76.6~81.4 | 7.2 |
20 | 88.2~96.5 | 7.2 | |
50 | 86.3~97.1 | 5.2 | |
鸡肝 | 10 | 87.6~105.5 | 6.0 |
20 | 78.9~92.3 | 5.9 | |
50 | 76.0~91.1 | 5.9 |
左 旋咪唑回收率为71.3%~105.5%,相对标准偏差RSD为5.2%~8.1%。本方法精密度和回收率都符合方法学的要求,说明本研究方法准确并具有可行性。
3 结论
根据左旋咪唑目标化合物的相关物理化学性质以及动物肝脏样品基质的特点,样品经乙腈-冰乙酸(1%)溶液分散固相萃取,经过无水硫酸钠(Na2SO4)及氨基(NH2)净化,采用C18色谱柱使其将目标化合物与杂质进行有效分离,选择多反应监测(MRM)扫描模式进行检测,以外标法定量。实验结果表明,左旋咪唑在质量浓度10~200 μg/L范围内具有良好的线性关系,相关系数为0.9996,方法定量检测限为10.0 μg/kg。左旋咪唑在猪肝、羊肝、牛肝、鸡肝4种样品中低、中、高3级水平添加下的加标回收率在71.3%~105.5%范围,相对标准偏差(n=6)为5.2%~8.1%。该检测方法在前处理、试剂消耗、检测速度方面明显优于传统的检测方法,用于检测动物肝脏中左旋咪唑的实际应用中取得良好的效果,为快速检测动物肝脏中左旋咪唑含量的标准制定提供技术支持。
【该文经CNKI学术不端文献检测系统检测,总文字复制比为4.7%。】
参考文献
[ 1]赵文成, 吕金国, 杨维仁,等.左旋咪唑在兽医临床中的应用[J].养殖技术顾问, 2011, 2: 160.
[2] Cholifah S, Kartinasari W F, Indrayanto G. J Liq Chromatogr Re-lat Technol, 2008, 31(2): 281.
[3]尹荣兰,沈国顺. 左旋咪唑研究进展[J].中国兽医杂志, 2005, 41 (5): 45-46.
[4] National Food Safety Information Center. National Food Safety Re-source Database/Pesticide and Veterinary Drugs/Limits of Pesti-cide and Veterinary Drugs/Levamisole(国家食品安全信息中心. 中国食品安全资源数据库/农兽药数据库/农兽药的限量/左旋咪唑).(2008-05-30). http://www.fsr.org.cn/User_Log.asp.
[5] El-Kholy H , Kemppainen B W. J Chromatogr B, 2003, 796: 371.
[6] Dreassi E, Corbini G, La Rosa C, et al. J Agric Food Chem, 2001, 49: 5702.
[7]林维宣,田苗,隋凯,等.HPLC法检测乳制品中左旋咪唑[J].中国乳品工业, 2001, 29(1): 24-26.
[8]曾勇,金秀娥,卢芳,等. 猪鸡组织中左旋咪唑残留的HPLC检测方法研究[J].中国兽医杂志, 2010, 44( 8) :17-21.
[9] 辛丹敏,周光明,黄成 ,等. 基质固相分散-高效液相色谱分析奶 粉中残留的左旋咪唑和甲苯咪唑[J].分析试验室, 2008, 27(S) : 365-367.
[10] Cherlet M, De Baere S, Croubels S, et al. J Chromatogr B, 2000, 742: 283.
[11]蔡伟群.用GC法测定盐酸左旋咪唑在驱虫药中的含量[J].亚太传统医学, 2009, 1: 24-25.
[12] De Baere S, Cherlet M, Croubels S, et al. Anal Chim Acta, 2003, 483:215.
(文章类别:CPST-C)
