





CopyRight 2009-2020 © All Rights Reserved.版权所有: 中国海关未经授权禁止复制或建立镜像

QuEChERS结合UPLC-MS/MS法测定畜禽肉中的硝基咪唑类药物
作者:姚璇 王波 刘兰霞 白兴斌 吕蓓蓓 周红艳 解迎双
姚璇 王波 刘兰霞 白兴斌 吕蓓蓓 周红艳 解迎双
Abstract Employing SHISEIDO C18 column (100 mm×2.1 mm, 2.7 μm), pure water-methanol was used as the mobile phase to analyze the components to be tested. QuEChERS method was selected as the sample pretreatment method for purification. By comparing the separating effects of different types of chromatographic column for target compounds, SHISEIDO C18 Column had a good effect on the separation of the target compounds. The methodological results showed that the target compounds had a good linear correlation within the range of 0.5-20 μg/L; correlation coefficients (r2) were all≥0.9976; and the detection limits of the method were 0.5 μg/kg and the quantative limit was 2.0 μg/kg. At three different concentration spiking levels, the average recoveries of nitroimidazole compounds in 6 kinds of sample matrix was 79.86%-113.8%. Chromatographic separation for 32 kinds of target compounds was achieved through investigating different chromatographic conditions and multiple mass spectrum conditions of instrument conditions. Practical sample verification proved that this method was simple, reliable, accurate, efficient and fast and had practical application value. Therefore, this method can be used for high-throughput qualitative screening and quantitative analysis of multiple nitroimidazole compounds in chicken, beef, pork and mutton.
Keywords liquid chromatography-tandem mass spectrometer; nitroimidazole compounds; poultry meat
硝基咪唑化合物作为一类抗原虫和抗极性很强的药物,被广泛用于禽畜等动物的饲养繁殖领域[1-3]。该类化合物不但具有动物致癌毒性,还存在细胞诱变性等潜在的、严重的生物安全问题,国际上许多国家及组织已相继出台法律法规,禁止在动物源性食品中使用硝基咪唑类化合物[4-5]。
近几年来,有关硝基咪唑类化合物残留的检验方法较多[6-7]。其中,采用液相色谱-串联质谱法对硝基咪唑类化合物进行检测时,特异性好、灵敏度高、稳定性好[8-10],目前已成为硝基咪唑类化合物检测的常用手段。在硝基咪唑类药物残留检测的样品预处理环节,常用的净化手段主要有固相萃取净化法、分子印迹净化法、QuEChERS等[11]。QuEChERS技术能够克服固相萃取净化法溶剂消耗量大、使用成本高,以及分子印迹净化法制备时间长的缺陷[12],在多种类兽药残留分析的样品前处理过程中得到了广泛应用。同时,该净化手段高速度、高效率、操作简便、成本较低,可实现高通量检测[13]。
鉴于上述问题,本研究采用QuEChERS技术结合高效液相色谱三重四级杆质谱仪建立了测定羊肉、牛肉、猪肉、鸡肉等畜禽肉中多种硝基咪唑类药物及其代谢物残留的方法。
1 实验部分
1.1 仪器与设备
Quattro Premier XE超高效液相色谱-串联质谱仪(Waters公司);ME55/02电子天平(梅特勒托利多公司);混旋仪(Heidolph公司);H2500R高速台式冷冻离心机(湖湘仪实验室仪器开发有限公司);SPeedVacTM SRF 110制冷型离心式真空浓缩仪(赛默飞世尔科技公司)。
1.2 试剂与材料
哌莫硝唑、羟基甲硝唑、米索硝唑、奥硝唑、左旋奥硝唑、依他硝唑(纯度>99%)、羟基二甲硝咪唑、甲硝唑、替硝唑、塞克硝唑、特尼达唑、羟基异丙硝唑、异丙硝唑、苄硝唑、非昔硝唑、帕硝唑、卡硝唑、特硝唑、罗硝唑、2-甲硝咪唑、地美硝唑、氯甲硝咪唑、苯硝咪唑、4-硝基咪唑、二甲硝唑、洛硝哒唑、甲苯咪唑、甲硝咪唑、甲疏咪唑、苯并咪唑、沙曲硝唑、苯酰甲硝唑(纯度>98%)购自德国Dr.Ehrenstorfer公司。
氨水(色谱纯)购自美国Sigma-Aldrich公司;甲醇(色谱纯)购自美国Fisher公司;乙酸乙酯(色谱纯)、乙腈(色谱纯)均购自德国Merck公司;QuEChERS用净化填料(C18、PSA、GCB、MgSO4)购自美国安捷伦公司;超纯水(18.2 MΩ·cm)由本实验室纯水仪制备。乙腈饱和正己烷:取体积相同的乙腈和正己烷,混合后进行一定时间的静置后取上层。
1.3 标准溶液配置
标准储备液配置(浓度100 mg/L):精密称取哌莫硝唑、羟基甲硝唑、奥硝唑、左旋奥硝唑各0.0101 g、羟基二甲硝咪唑、甲硝唑、替硝唑、塞克硝唑、特尼达唑、羟基异丙硝唑、异丙硝唑、苄硝唑、非昔硝唑、帕硝唑、卡硝唑、米索硝唑、特硝唑、罗硝唑、2-甲硝咪唑、地美硝唑、氯甲硝咪唑、苯硝咪唑、4-硝基咪唑、二甲硝唑、洛硝哒唑、甲苯咪唑、甲硝咪唑、甲疏咪唑、苯并咪唑、依他硝唑、沙曲硝唑、苯酰甲硝唑各0.0102 g(均精确至0.0001 g),用甲醇溶解后,定容于100 mL容量瓶内,配制成质量浓度为100 mg/L的单标准储备溶液,于-18℃的冰箱中保存6个月。
混合标准中间液:分别吸取32个硝基咪唑类化合物的单个标准储备液(100 mg/L)100 μL于100 mL容量瓶中,后用甲醇进行定容,配制质量浓度为1 mg/L的混合标准中间液,在4℃的冰箱中保存。
1.4 样品采集与制备
猪肉、牛肉、羊肉、鸡肉经刀式研磨仪均质后,于-20℃冷冻保存,待用。
1.5 样品前处理
精确称取5.00±0.01 g制备后的1.4所述待测样品于50 mL带有密封盖的管状试样容器中,加入10 mL乙酸乙酯后立即涡旋10 min后超声10 min,在4℃、10000 r/min条件下离心5 min,将上清液移至平行蒸发瓶内,向带有残渣的管状试样容器中再次加入10 mL乙酸乙酯,重复提取后合并提取液,平行蒸发至近干,用2 mL 2%甲酸水(体积分数)涡旋溶解,待净化。
向待净化液中加入乙腈饱和正己烷2 mL,涡旋脱脂1 min,静止5 min,弃去正己烷层,依次加入50 mg PSA、50 mg C18,涡旋混匀1 min,在4℃、10000 r/min条件下离心5 min,上清液过0.22 μm滤膜,待检测。
1.6 仪器条件
1.6.1 色谱条件
使用SHISEIDO C18色谱柱(2.7 μm,100 mm×2.1 mm),柱温为30℃,进样量为2 μL,流速为0.30 mL/min,流动相A为纯水,流动相B为甲醇,液相洗脱程序见表1。
表1 超高效液相色谱梯度洗脱程序
Table 1 UPLC gradient elution program
时间 (min) | 流速 (mL/min) | A (%) | B (%) |
0 | 0.300 | 2.0 | 98.0 |
5 | 0.300 | 10.0 | 90.0 |
15 | 0.300 | 30.0 | 70.0 |
20 | 0.300 | 40.0 | 60.0 |
21 | 0.300 | 40.0 | 60.0 |
26 | 0.300 | 90.0 | 10.0 |
28 | 0.300 | 5.0 | 95.0 |
30 | 0.300 | 5.0 | 95.0 |
1.6.2 质谱测定条件
离子源为ESI源正离子模式,扫描方式为多反应监测(MRM)条件下的分段采集,Capillary voltage电压为3.0 kV,离子源温度为120℃,Desolvation温度为350℃,反吹气流速为50 L/Hr,其他MRM参数见表2。
2 结果与讨论
2.1 色谱条件的选择
根据文献中报道的硝基咪唑类化合物中等极性的特点[14-16],本研究比较了SHISEIDO C18色谱柱和Nanologica SVEA C18色谱柱对目标化合物的分离效果。Nanologica SVEA C18色谱柱填料对目标物的保留能力弱,峰形较宽,且甲硝唑和羟基甲硝唑不能得到分离。增加SHISEIDO C18(100 mm×2.1 mm,2.7 μm)色谱柱柱长使得目标物流出时间延后,与不保留的杂质进行了更好地分离,保留能力好,峰形对称相对更优,可以更好地与共流干扰物得到分离,因此最终选择使用SHISEIDO C18(100 mm×2.1 mm,2.7 μm)色谱柱。
本研究还比较了0.1%甲酸水(V/V)-甲醇、含5 mmol/L乙酸铵-甲醇和纯水-甲醇3个流动相体系对硝基咪唑类药物及其代谢物分离效果的影响。当流动相体系通过加入甲酸调节pH值后,甲硝唑、羟基甲硝唑在1 min内出峰。在实际样品测定过程中,因甲硝唑、羟基甲硝唑出峰时间较早而无法与杂质进行较好分离,因此易受基质干扰。当溶液中加入乙酸铵后,在对帕硝唑、苄硝唑两种咪唑类化合物进行检测的过程中,发现其色谱峰出现明显的峰展宽的现象,不仅影响了上述2种化合物的灵敏度,也给这2种化合物的定量带来了一定的困难。因此,本研究选用SHISEIDO C18色谱柱,纯水-甲醇作为流动相分离硝基咪唑类药物及其代谢物,使其能够较好地实现分离。
2.2 样品前处理条件的优化
2.2.1 提取条件的优化
硝基咪唑类化合物的提取试剂一般为乙酸乙酯、乙腈、二氯甲烷、酸性乙腈和碱性乙腈等[17-18],本研究对比了采用乙酸乙酯、乙腈、二氯甲烷、0.1%甲酸乙腈(V/V)、0.1%氨水乙腈(V/V)时硝基咪唑类化合物的提取效率(图1)。乙腈作为提取溶剂时,硝基咪唑类化合物的整体回收率在23.21%~68.42%。二氯甲烷作为提取溶剂时,沙曲硝唑、特硝唑、罗硝唑、奥硝唑、左奥硝唑、地美硝唑、苯硝咪唑的回收率<20%。0.1%甲酸乙腈(V/V)、0.1%氨水乙腈作为提取溶液时,硝基咪唑类化合物的整体回收率在62.43%~91.26%。乙酸乙酯作为提取溶液时,硝基咪唑类化合物的整体回收率在79.86%~113.8%之间。因此,本研究最终选择乙酸乙酯作为提取溶剂。
2.2.2 净化条件的优化
本研究所涉及的样品基质均含有大量的脂肪[19-20],在样品的前处理过程中有效除去脂肪从而降低基质效应是关键。以各硝基咪唑类化合物的回收率为考核指标,通过实验分别考察正己烷、乙腈饱和正己烷、石油醚、环己烷4种溶剂对6种不同基质提取液的脱脂净化效果。采用正己烷、石油醚、环己烷进行脱脂实验,非昔硝唑的回收率只有40%,而乙腈饱和正己烷对硝基咪唑类化合物的整体回收率在79.86%~113.8%之间,且净化效果较好,因此本研究的脱脂溶剂最终选择乙腈饱和正己烷。
QuEChERs技术利用净化剂填料与样品基质中杂质相互作用的原理,通过对杂质进行吸附达到净化目的[21-22]。因此,在QuEChERs技术净化过程中一个影响检测回收率的最重要参数就是吸附剂的选择。C18可有效吸附样品中的非极性成分,起到去除杂质的作用;石墨化炭黑(GCB)能够通过吸附有色物质和固醇类化合物达到净化作用。PSA其主要成分乙二胺-N-丙基硅烷能高效吸附样品中有机酸、酚类化合物,可以达到去除基质中相关干扰物的目的。本研究对C18、GCB、PSA吸附剂对动物源性样品的净化作用进行了比对。结果显示,GCB对目标化合物具有一定的吸附作用,添加后目标化合物的回收率的稳定性较差,回收率<50%;将PSA和C18以一定比例混合后作为净化剂,目标物的回收率范围为79.86%~113.8%(图2),因此实验最终选用PSA和C18作为实验过程中的净化剂。
3 方法学评估
3.1 标准曲线、检出限、定量限的确定
将硝基咪唑类化合物的1000 μg/L标准溶液按梯度稀释,梯度浓度分别为0.5 μg/L、1.0 μg/L、3.0 μg/L、5.0 μg/L、10.0 μg/L、15.0 μg/L、20.0 μg/L。采用1.6所述仪器检测条件进行测定,使用外标法进行定量。以硝基咪唑类化合物的梯度浓度值为横坐标,其定量离子的峰面积响应值为纵坐标,绘制本实验条件下硝基咪唑类化合物在梯度浓度(0.5~20 μg/L)范围内的线性曲线。结果如表3所示,在0.5~20 μg/L范围内各硝基咪唑类药物及其代谢物具有良好的线性相关性,r2≥0.9976。
本研究采用经检测后空白的鸡肉、猪肉、羊肉、牛肉样品,分别添加0.1 μg/kg、0.5 μg/kg、1.0 μg/kg、2.0 μg/kg、3.0 μg/kg的32种混合标准溶液(n = 5),经前处理后上机检测,根据S/N≥3倍信噪比下各硝基咪唑类化合物色谱峰的响应值,得出畜禽肉中硝基咪唑类化合物的方法检出限为0.5 μg/kg。根据10倍信噪比下目标化合物色谱峰的响应值,得出畜禽肉中硝基咪唑类化合物的方法定量限为2.0 μg/kg。
3.2 回收率与精密度
准确称取经检测后不含有硝基咪唑类化合物的鸡肉、猪肉、羊肉、牛肉空白基质样品各2.0 g,设定3.0 μg/kg、6.0 μg/kg和10.0 μg/kg 3个添加水平,每组做6个平行样品,按照1.5所述样品预处理方法和1.6所述仪器条件测定本研究的回收率和相对标准偏差。如表3所示,将4种基质中硝基咪唑类化合物的回收率和精密度进行平均值计算,平均回收率为79.86%~113.8%,平均相对标准偏差为2.9%~8.2%(n = 6)。
4 实际样品测定
使用该方法对超市购买的鸡肉、猪肉、牛肉、羊肉共30批市售样品进行测定,各采样样品中均未检出硝基咪唑类化合物。
5 结论
本研究建立了QuEChERS结合UPLC-MS/MS法测定畜禽肉中的硝基咪唑类化合物的方法。针对鸡肉、牛肉、猪肉、羊肉样品的基质特点,对提取条件、净化剂种类、脱脂溶剂等样品预处理参数进行了优化。通过方法学考察,在3个不同浓度水平下,各硝基咪唑类化合物在6种基质样品中的平均回收率为79.86%~113.8%,精密度为2.9%~8.2%。通过对仪器条件的色谱参数、质谱参数进行考察,建立了对32种目标化合物的色谱分离方法。相对于固相萃取法,本研究建立的净化方法具有操作简单、耗时短、溶剂消耗量小等特点,可用于鸡肉、牛肉、猪肉、羊肉中多种硝基咪唑类化合物的高通量定性、定量分析。
参考文献
[1] Wang C, Chen M, Hu Q, et al. Non-lethal microsampling and rapid identification of multi-residue veterinary drugs in aquacultured fish by direct analysis in real time coupled with quadrupole-Orbitrap high-resolution mass spectrometry[J]. Microchemical Journal, 2021, 160(13): 1-8.
[2] Kawamura M. Simple and rapid screening for psychotropic naturalproducts using direct analysis in real time (DART)-TOFMS[J]. Yakugaku Zasshi, 2009, 129(6): 719.
[3] Jagerdeo E, Abdel Rehim M. Screening of cocaine and its metabo-lites in human urine samples by direct analysis in real-time source coupled to time-of-flight mass spectrometry after online preconcentration utilizing microextraction by packed sorbent[J]. Am SocMass Spectrom, 2009, 20: 719-725.
[4] Chernetsova E S, Morlock G E, Revelsky I A. DART mass spectrometry and its applications in chemical analysis[J]. Russian Chemical Reviews, 2011, 80(36): 249-271.
[5] Yang H M, Wan D B, Song F R, et al. Argon direct analysis in real time mass spectrometry in conjunction with makeup solvents: a method for analysis of labile compounds[J]. Analytical Chemistry, 2013, 85(3): 1305-1309.
[6] Dane A J, Cody R B. Selective ionization of melamine in powdered milk by using argon direct analysis in real time (DART) mass spectrometry[J]. Analyst, 2010, 135(4): 696-699.
[7] Yu S X, Crawford E, Tice J, et al. Bioanalysis without sample cleanup or chromatography: the evaluation and initial implementation of direct analysis in real time ionization mass spectrometry for the quantification of drugs in biological matrixes[J]. Analytical Chemistry, 2008, 81(1): 193-202.
[8] Vaclavik L, Cajka T, Hrbek V, et al. Ambient mass spectrometry employing direct analysis in real time (DART) ion source for olive oil quality and authenticity assessment[J]. Analytica Chimica Acta, 2009, 645(1): 56-63.
[9] Wang L, Zhao P Y, Zhang F Z, et al. Direct analysis in real time mass spectrometry for the rapid identification of four highly hazardous pesticides in agrochemicals[J]. Rapid Communications in Mass Spectrometry, 2012, 26(16): 1859-1867.
[10] Hajslova J, Cajka T, Vaclavik L, Challenging applications offered by direct analysis in real time (DART) in food-quality and safety analysis[J]. Trac Trends in Analytical Chemistry, 2011, 30(2): 204-218.
[11] Newsome GA, Ackerman LK, Johnson KJ. Humidity affects relativeion abundance in direct analysis in real time mass spectrometry of hexamethylene triperoxide diamine [J]. Analytical Chemistry, 2014, 86(24): 11977-11980.
[12] Zhou Z G, Zhang J L, Zhang W, et al. Rapid screening for synthetic antidiabetic drug adulteration in herbal dietary supplements using direct analysis in real time mass spectrometry[J]. Analyst, 2011, 136(12): 2613-2618.
[13] Hajslova J, Cajka T, Vaclavik L. Challenging applications offered by direct analysis in real time (DART) in food-quality and safety analysis[J]. Trac Trends in Analytical Chemistry, 2011, 30(2): 204-218.
[14]蓝草, 邵琳智, 徐娟. 实时直接分析-静电场轨道阱高分辨质谱法应用于化妆品中 15 种抗过敏药物的定性和定量分析[J]. 理化检验(化学分册), 2020, 56(6): 686-691.
[15]韩晔华, 欧阳萍, 张艳芬, 等. 基于实时直接分析质谱技术的氯铝酸及其复合离子液体分析[J]. 中国科学: 化学, 2020, 50(6): 720-728.
[16]蓝草, 邵琳智, 陈思敏. 实时直接分析高分辨质谱法快速检测化妆品中的氯霉素[J]. 分析测试学报, 2019, 38(12): 1503-1506.
[17]王晓利, 郭涛, 苑金鹏, 等. 高效液相色谱-串联质谱法测定蔬菜中喹诺酮类抗生素残留[J]. 分析试验室, 2019, 38(8): 916-920.
[18]程志, 宿书芳, 魏莉莉, 等. 通过式固相萃取净化-液相色谱-串联质谱法测定豆芽中10种喹诺酮类抗生素[J]. 分析试验室, 2020, 39(2): 131-136.
[19]宋晓婉, 梁先龙, 冯娟, 等. QuECHERS方法检测动物源性食品中15种喹诺酮类药物残留量及风险监测研究[J]. 食品安全导刊, 2019(18): 89-91.
[20] Maurer Christine K, Steinbach Anke, Hartmann Rolf W. Development and validation of a UHPLC-MS/MS procedure for quantification of the Pseudomonas Quinolone Signal in bacterial culture after acetylation for characterization of new quorum sensing inhibitors[J]. Journal of pharmaceutical and biomedical analysis, 2013, 86(3): 176-182.
[21]卢剑, 寻知庆, 汪晨霞, 等. 高效液相色谱串联质谱法测定化妆品中15种喹诺酮类抗生素[J]. 日用化学工业, 2019, 49(6): 403-409.
[22]史艳艳, 高琳, 李俊, 等. 液相色谱-串联质谱法检测饲料中洛美沙星、培氟沙星、氧氟沙星、诺氟沙星残留[J]. 食品安全质量检测学报, 2019, 10(11): 3367-3375.
表2 硝基咪唑类化合物的质谱参数
Table 2 MS/MS parameters of NMZs
化合物名称 | (m/z) | (m/z) | (V) | (eV) | 离子源 | 化合物名称 | (m/z) | (m/z) | (V) | (eV) | 离子源 |
哌莫硝唑 | 255.08 | 98.37 | 30 | 20 | ESI+ | 奥硝唑 | 220.0 | 128.1* | 28 | 22 | ESI+ |
124.39* | 15 | 82.1 | 22 | ||||||||
羟基甲硝唑 | 187.78 | 123.14 | 30 | 25 | ESI+ | 左旋奥硝唑 | 220.0 | 128.1* | 26 | 22 | ESI+ |
126.17* | 30 | 82.1 | 22 | ||||||||
羟基二甲硝咪唑 | 157.93 | 55.46 | 28 | 25 | ESI+ | 2-甲硝咪唑 | 128.0 | 82.2 | 30 | 18 | ESI+ |
94.34* | 25 | 111.1* | 16 | ||||||||
甲硝唑 | 172.31 | 82.13 | 25 | 25 | ESI+ | 地美硝唑 | 142.0 | 96.1* | 35 | 56 | ESI+ |
127.99* | 10 | 81.0 | 75 | ||||||||
替硝唑 | 247.88 | 82.32 | 30 | 30 | ESI+ | 氯甲硝咪唑 | 162.0 | 116.1* | 32 | 20 | ESI+ |
128.27* | 10 | 145.0 | 20 | ||||||||
塞克硝唑 | 186.19 | 82.32 | 30 | 20 | ESI+ | 苯硝咪唑 | 164.0 | 118.1* | 28 | 16 | ESI+ |
128.27* | 25 | 91.1 | 16 | ||||||||
特尼达唑 | 186.19 | 82.32 | 25 | 25 | ESI+ | 4-硝基咪唑 | 114.0 | 97.0* | 26 | 15 | ESI+ |
128.24* | 20 | 84.0 | 14 | ||||||||
羟基异丙硝唑 | 185.95 | 122.32 | 30 | 20 | ESI+ | 二甲硝唑 | 142.0 | 96.0* | 24 | 18 | ESI+ |
168.25* | 20 | 81.0 | 25 | ||||||||
异丙硝唑 | 169.89 | 109.18 | 35 | 20 | ESI+ | 洛硝哒唑 | 201.0 | 140.1* | 26 | 15 | ESI+ |
124.18* | 20 | 110.1 | 15 | ||||||||
苄硝唑 | 261.07 | 91.35 | 25 | 25 | ESI+ | 甲苯咪唑 | 296.1 | 264.1* | 28 | 15 | ESI+ |
107.36* | 15 | 236.0 | 18 | ||||||||
非昔硝唑 | 233.10 | 93.39 | 30 | 15 | ESI+ | 甲硝咪唑 | 128.0 | 111.0* | 27 | 12 | ESI+ |
106.31* | 25 | 98.0 | 18 | ||||||||
帕硝唑 | 233.04 | 93.33 | 25 | 10 | ESI+ | 甲疏咪唑 | 115.0 | 88.0* | 30 | 12 | ESI+ |
106.31* | 25 | 83.0 | 15 | ||||||||
卡硝唑 | 245.07 | 75.28 | 20 | 10 | ESI+ | 苯并咪唑 | 164.0 | 118.0* | 25 | 25 | ESI+ |
118.29* | 20 | 91.0 | 30 | ||||||||
米索硝唑 | 202.19 | 145.30 | 30 | 25 | ESI+ | 依他硝唑 | 215.1 | 112* | 30 | 25 | ESI+ |
174.22* | 15 | 104.1 | 22 | ||||||||
特硝唑 | 186.1 | 128.0* | 28 | 15 | ESI+ | 沙曲硝唑 | 290.2 | 128.0* | 30 | 25 | ESI+ |
82.1 | 12 | 82.1 | 22 | ||||||||
罗硝唑 | 201.1 | 140.0* | 32 | 18 | ESI+ | 苯酰甲硝唑 | 275.26 | 264.1* | 30 | 23 | ESI+ |
181.1 | 15 | 91.1 | 26 |
注: “*”为定量离子
表2(续)
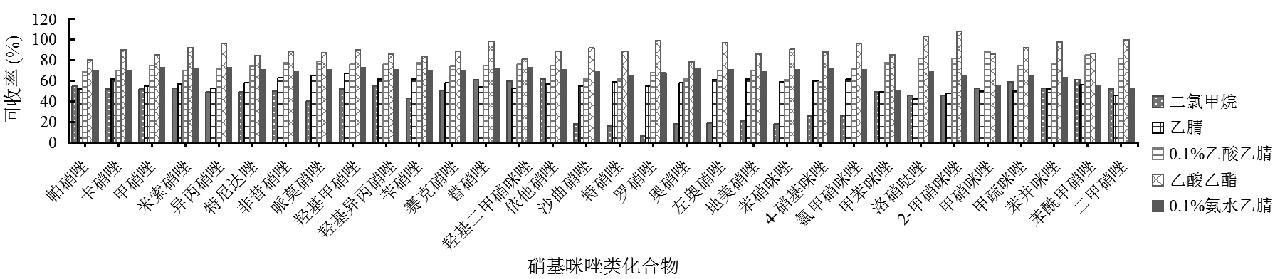
图1 硝基咪唑化合物在不同溶剂下的提取效率图
Fig.1 Extraction efficiency plot of nitroimidazole compounds under different solvents
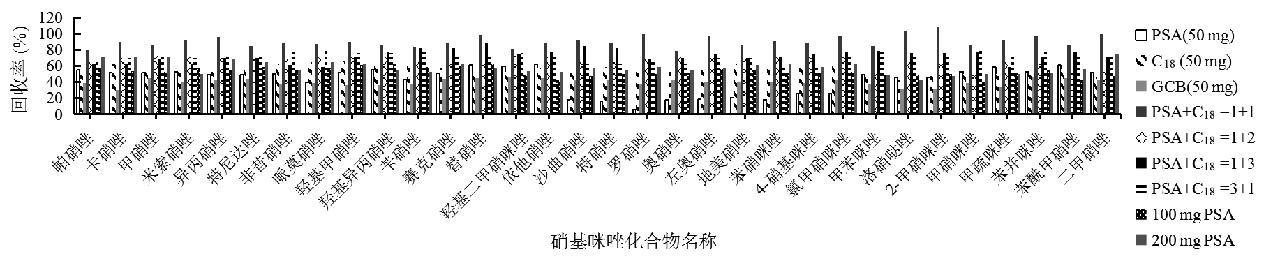
图2 硝基咪唑化合物在不同吸附剂下的提取效率图
Fig.2 Extraction efficiency plot of nitroimidazole compounds under different adsorbents
表3 6种硝基咪唑类化合物方法学考察数据表
Table 3 Data sheet for methodological investigation of six nitroimidazole compounds
化合物 | (μg/L) | r2 | (μg/kg) | (μg/kg) | 添加量 3.0 μg/kg | 添加量 6.0 μg/kg | 添加量 10.0 μg/kg | |||||
回收率 (%) | RSD (%) | 回收率 (%) | RSD (%) | 回收率 (%) | RSD (%) | |||||||
呱漠硝唑 | 0.50~20.0 | 0.9995 | 0.5 | 2.0 | 87.82 | 4.80 | 85.41 | 3.60 | 88.37 | 3.10 | ||
羟基甲硝唑 | 0.50~20.0 | 0.9994 | 0.5 | 2.0 | 91.02 | 3.80 | 90.24 | 3.70 | 91.79 | 4.10 | ||
羟基二甲硝咪唑 | 0.50~20.0 | 0.9996 | 0.5 | 2.0 | 92.30 | 3.50 | 88.90 | 3.40 | 89.50 | 3.60 | ||
甲硝唑 | 0.50~20.0 | 0.9999 | 0.5 | 2.0 | 94.55 | 4.80 | 85.90 | 3.60 | 106.0 | 3.50 | ||
替硝唑 | 0.50~20.0 | 0.9994 | 0.5 | 2.0 | 98.02 | 3.90 | 97.10 | 4.30 | 95.60 | 3.40 | ||
塞克硝唑 | 0.50~20.0 | 0.9994 | 0.5 | 2.0 | 89.25 | 3.50 | 87.14 | 4.70 | 90.47 | 3.60 | ||
特尼达唑 | 0.50~20.0 | 0.9998 | 0.5 | 2.0 | 92.85 | 3.20 | 80.23 | 4.80 | 90.56 | 3.10 | ||
羟基异丙硝唑 | 0.50~20.0 | 0.9996 | 0.5 | 2.0 | 96.45 | 3.70 | 99.20 | 3.40 | 96.20 | 3.60 | ||
异丙硝唑 | 0.50~20.0 | 0.9994 | 0.5 | 2.0 | 95.57 | 3.00 | 90.80 | 3.80 | 99.50 | 3.30 | ||
苄硝唑 | 0.50~20.0 | 0.9998 | 0.5 | 2.0 | 98.72 | 3.80 | 99.54 | 4.60 | 92.30 | 4.10 | ||
非昔硝唑 | 0.50~20.0 | 0.9993 | 0.5 | 2.0 | 94.22 | 3.80 | 94.20 | 3.70 | 97.76 | 3.10 | ||
帕硝唑 | 0.50~20.0 | 0.9994 | 0.5 | 2.0 | 93.52 | 3.50 | 92.90 | 3.40 | 90.60 | 3.60 | ||
卡硝唑 | 0.50~20.0 | 0.9990 | 0.5 | 2.0 | 94.54 | 3.80 | 92.30 | 4.60 | 106.0 | 3.50 | ||
米索硝唑 | 0.50~20.0 | 0.9994 | 0.5 | 2.0 | 92.80 | 2.90 | 92.80 | 3.30 | 113.8 | 4.40 | ||
依他硝唑 | 0.50~20.0 | 0.9984 | 0.5 | 2.0 | 89.25 | 3.50 | 97.14 | 3.70 | 103.6 | 4.60 | ||
沙曲硝唑 | 0.50~20.0 | 0.9982 | 0.5 | 2.0 | 92.85 | 3.20 | 90.23 | 8.20 | 90.56 | 8.10 | ||
特硝唑 | 0.50~20.0 | 0.9990 | 0.5 | 2.0 | 96.45 | 4.70 | 99.20 | 3.40 | 96.20 | 3.60 | ||
苯酰甲硝唑 | 0.50~20.0 | 0.9994 | 0.5 | 2.0 | 95.57 | 3.00 | 90.80 | 3.80 | 99.50 | 3.30 | ||
苯并咪唑 | 0.50~20.0 | 0.9992 | 0.5 | 2.0 | 98.66 | 5.90 | 97.10 | 4.50 | 87.10 | 3.20 | ||
罗硝唑 | 0.50~20.0 | 0.9976 | 0.5 | 2.0 | 95.88 | 4.30 | 96.50 | 4.00 | 87.90 | 8.20 | ||
甲疏咪唑 | 0.50~20.0 | 0.9995 | 0.5 | 2.0 | 95.54 | 4.70 | 97.16 | 3.90 | 95.43 | 3.40 | ||
甲硝咪唑 | 0.50~20.0 | 0.9984 | 0.5 | 2.0 | 94.55 | 4.80 | 85.90 | 3.60 | 106.0 | 3.50 | ||
奥硝唑 | 0.50~20.0 | 0.9994 | 0.5 | 2.0 | 98.02 | 3.90 | 97.10 | 4.30 | 95.60 | 3.40 | ||
左奥硝唑 | 0.50~20.0 | 0.9999 | 0.5 | 2.0 | 89.25 | 3.50 | 87.14 | 3.70 | 90.47 | 4.60 | ||
2-甲硝咪唑 | 0.50~20.0 | 0.9989 | 0.5 | 2.0 | 92.85 | 3.20 | 80.23 | 3.80 | 90.56 | 6.10 | ||
地美硝唑 | 0.50~20.0 | 0.9982 | 0.5 | 2.0 | 96.45 | 3.70 | 99.20 | 3.40 | 96.20 | 3.60 | ||
氯甲硝咪唑 | 0.50~20.0 | 0.9993 | 0.5 | 2.0 | 92.33 | 5.50 | 90.15 | 3.80 | 93.26 | 7.30 | ||
苯硝咪唑 | 0.50~20.0 | 0.9994 | 0.5 | 2.0 | 88.69 | 3.10 | 87.50 | 3.20 | 89.75 | 3.20 | ||
4-硝基咪唑 | 0.50~20.0 | 0.9996 | 0.5 | 2.0 | 79.86 | 6.30 | 81.07 | 3.70 | 84.29 | 4.50 | ||
二甲硝唑 | 0.50~20.0 | 0.9996 | 0.5 | 2.0 | 98.66 | 5.90 | 97.10 | 4.50 | 87.10 | 3.20 | ||
洛硝哒唑 | 0.50~20.0 | 0.9996 | 0.5 | 2.0 | 96.33 | 4.60 | 92.50 | 3.50 | 97.20 | 4.60 | ||
甲苯咪唑 | 0.50~20.0 | 0.9995 | 0.5 | 2.0 | 88.72 | 3.80 | 89.54 | 3.60 | 92.30 | 4.10 | ||
表3(续)
