





CopyRight 2009-2020 © All Rights Reserved.版权所有: 中国海关未经授权禁止复制或建立镜像

建设基于下一代测序技术( NGS)的传染病监测预警平台构想
作者:潘衍宇 竺文杰 金东新 刘曦宇 张俊波
潘衍宇 竺文杰 金东新 刘曦宇 张俊波
摘 要 我国海关坚持“口岸疫情防控海关必坚守”要求,身处“外防输入”的第一线,严格落实“多病同防”工作,亟需既能进行深度病原识别及精准溯源,又能承担未来口岸常规传染病监测的统一化实验室监测预警平台。下一代测序技术为口岸在传染病监测提供了新的借鉴思路。本文介绍了若干全球范围内传统及新型的传染病监测预警平台架构及优缺点,并提出了组建基于下一代测序技术的海关系统新型传染病监测预警实验室平台的一些设计构想,为科学精准做好口岸疫情防控工作,实现精准检疫提供技术性参考。
关键词 全基因组测序;宏基因组测序;传染病监测预警;海关
The Establishment of an Early Warning Platform for Infectious Disease Surveillance Based
on Next- Generation Sequencing Technology
PAN Yan-Yu 1 ZHU Wen-Jie 1 JIN Dong-X in 2 L IU X i -Yu 2 ZHANG Jun-Bo 1*
Abstract In recent years, serving as the front-line defense against imported cases, China Customs adheres to the requirement of “the Customs must stand firm in preventing and controlling epidemics at ports”, and strictly implement the task of “multi-disease prevention”. There is an urgent need for a unified laboratory monitoring and early warning platform that can not only conduct in-depth pathogen identification and precise tracing, but also undertake routine monitoring of infectious diseases at ports in the future. The rise and application of Next Generation Sequencing (NGS) technology in the field of early warning provides a new perspective for infectious disease surveillance at customs ports. This article introduces the architecture, advantages and disadvantages of traditional and new infectious disease monitoring and early warning platforms around the world, and puts forward some design thoughts for the establishment of a new platform within the customs system based on NGS, aiming to provide reference for accurate prevention and control of epidemics at ports and realize precise quarantine.
Keywords whole genome sequencing; metagenomic Next-Generation Sequencing (mNGS); monitoring and early warning of infectious diseases; customs
基金项目:国家重点研发计划(2021YFC2301105)
第一作者:潘衍宇(1994—),男,汉族,浙江宁波人,硕士,主要从事卫生检疫及物流监管工作,E-mail: panda_mph@outlook.com
通信作者:张俊波(1984—),男,汉族,浙江宁波人,本科,主要从事卫生检疫及物流监管工作,E-mail: 291854396@qq.com
1. 镇海海关 宁波 315000
2. 宁波海关 宁波 315000
1. Zhenhai Customs, Ningbo 315000
2. Ningbo Customs, Ningbo 315000
国境口岸作为防控境外传染病输入的重要关口,承担着传染病防控的巨大压力。面对复杂多变的全球新发再发传染病态势,监测手段亟待转型升级,从而更好地满足当前海关系统精准检疫的需求。下一代测序技术在传染病监测预警领域的应用,为海关系统构建新型传染病监测预警平台提供了可能。
1 测序技术应用简述
下一代测序技术(Next Generation Sequencing,NGS)即第二代测序技术,是一种可同时对数百万DNA片段进行高通量测序的方法,能够快速准确地测出物种的全部遗传信息[1-2]。该测序方法可支持两种测序策略——全基因组测序(Whole Genome Sequencing,WGS)和宏基因组测序(Metagenomic Next Generation Sequencing,mNGS)。简要来说,WGS需要对样本进行分离培养,研究对象为具体某一纯菌株,序列质量较高,目标序列覆盖率高[2-3]。宏基因组测序不需要进行病原菌的分离培养,对经一定处理后的原始样本(如污水样本)直接测序,从而得到样品本身所携带的全部微生物遗传信息,但序列质量相对较低,目标序列覆盖率较低[4]。
随着基因测序技术的不断发展及测序成本的降低,传统的检验技术,包括脉冲场凝胶电泳(Ppulsed Field Ggel Electrophoresis,PFGE)、多位点序列分型(Multilocus Sequence Typing,MLST)、鉴定血清型的玻片凝集或多重PCR法等正在被NGS测序分析逐步替代,例如,美国疾病预防控制中心2018年起逐渐弃用PFGE,采用WGS技术作为食源性疾病鉴定及溯源的金标准[5-6]。再如,传统的MLST分型只利用了细菌全基因组中的7个管家基因序列进行分型,分辨率较低,难以单独用于暴发事件的溯源分析,目前多数实验室不再单独开展基于7个管家基因的PCR扩增进行菌株分型,而是基于分析NGS测序数据时顺便批量得出传统的分型结果,如血清型别、ST型别等[7-8]。
尽管多重PCR、微阵列或蛋白质组学等分析可在样本采集后数小时内提供病原体鉴定结果,但这些检测方法受限于对被检者受何种病原感染的提前预判,不能满足未来海关口岸防范众多不确定性的高危甚至未知病原的实际应用要求,同时也不能满足遭遇口岸输入性传染病暴发风险时进行精准溯源的要求。使用大规模并行NGS测序生成WGS或mNGS数据,可在约12 h内对样本中的数百种微生物进行全面分析,甚至有能力发现新型传染病病原[1, 3-4]。当前,基于NGS测序的病原菌识别及溯源相关技术已经非常成熟,特别是在新冠病毒溯源调查中得到充分的发展,世界范围出现大批应用实例[9-12]。这些都为海关系统构建以NGS测序技术为主的致病菌监测预警体系提供了科学可行的客观条件。
2 传统传染病监测预警系统概述
2.1 传统传染病监测预警系统
下面,笔者将介绍3种不同类型的传统监测预警平台。
2.1.1 ProMED-mail项目
新发疾病监测项目(The Program for Monitoring Emerging Diseases,ProMED-mail)是国际传染病学会(International Society for Infectious Diseases,ISID)于1994年建立的一套全球传染病监测报送系统,其成员由全球不同领域专家团队组成[13-14]。该项目通过在线平台和电子邮件免费向所有订阅者公开监测信息,其主要目的是尽快传递有关全球传染病暴发的最新情况,从而协助各国际组织、国家及地区尽早采取预防及控制措施。
该项目实时生成有关新发传染病、不明原因疾病暴发的报告,这些报告极有可能成为未来重大公共卫生事件的早期信号[13]。该系统在过去的30年间率先报道了包括SARS、中东呼吸综合征(MERS)、埃博拉等众多传染病暴发事件[15]。
该项目虽然发布环节有专门的信息审查小组进行审核,但由于其对提交信息的成员没有法规性质的约束力,这种基于个人或小组诚信提交的信息,其准确性存在潜在风险。另外,该种基于“大众”提交信息的模式,当扩大提交信息人员数量时,则信息质量良莠不齐,审核负担加重;当精减提交信息人员数量时,则无法全面获得各地重要公共卫生事件。该项目的定位更类似于各国政府官方传染病监测网络的良好补充,特别是在不明原因疾病的早期报道中可发挥一定作用[14]。
2.1.2 我国传染病自动预警信息系统
2008年中国疾病预防控制中心在国家法定传染病报告系统的基础上正式上架了一套子系统——国家传染病自动预警信息系统[16-17]。该子系统通过简便实用的固定阈值法及移动百分位数法实时分析全国法定传染病监测数据,若某地区报告病例数超出预警阈值时(依病种不同分为固定值与动态变化值两种),自动通过短信的形式将预警信息发送至具体区县的相关监测人员,简易流程如图1所示,该系统稳定运行至今已成功预防数千起传染病暴发扩散。
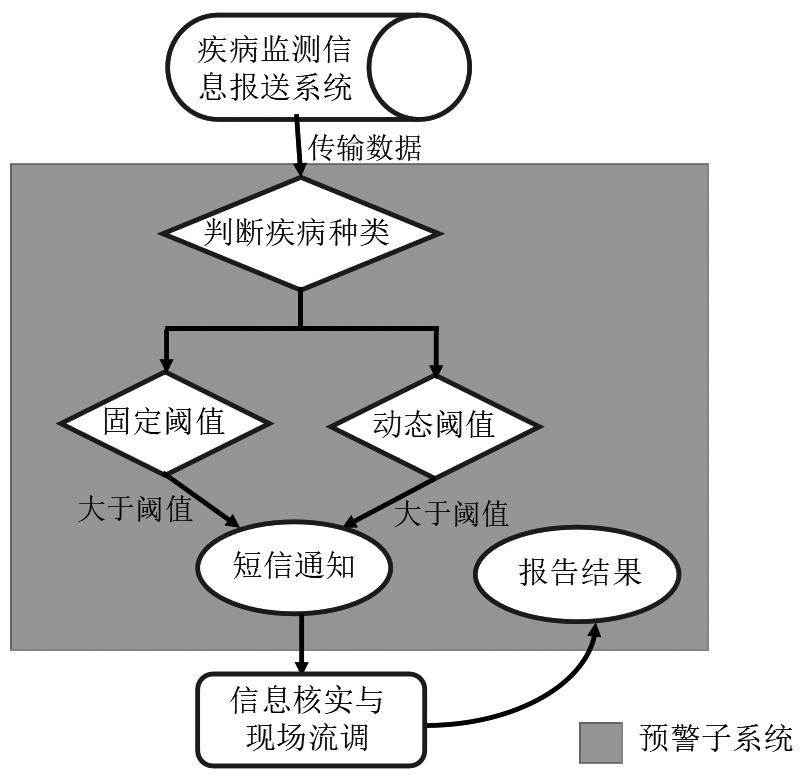
图1 国家传染病自动预警信息系统运行示意图
Fig.1 Schematic diagram of the operation of National Outbreak Automatic Detection and Response System
2.1.3 海关系统监测项目
2018年,海关总署成立全球传染病疫情信息工作组。前期根据世界卫生组织、美国疾控中心等官方网站通报信息建立了全球疫情信息数据库,同时采取网络爬虫等技术定期收集一系列指定国际组织、政府及科研机构的官方网站数据来实现疫情的每日动态监测。该系统另结合了地理信息系统实现了疫情数据的可视化。
以上3种模式,无论是专家组上报+邮件通报模式,还是哨点日常上报+自动监测预警模式,或自动爬取各监测机构通报数据+人工综合研判的方法,均属于传统的传染病监测预警模式,即收集事件的发生数量或率作为原始数据进行统计学的分析处理,从而做出判断。其结果只局限于统计学的推论,难以关联最直接的病原学证据。
2.2 基于NGS的传染病监测预警新模式
传统监测模式只知群体不知个人,由于无法关联到实验室证据,难以分辨出同时间或地区出现的阳性病例是否真实存在关联。当前基于NGS测序技术搭建的监测平台主要以致病菌的识别、溯源为主要目的。其可直接从最本质的基因组序列入手,做出有生物学证据的精准判断。下面介绍几种基于NGS测序技术的监测预警平台。
2.2.1 美国GalaxyTrakr项目
Galaxy为2005年开放的全球生物信息学共享服务器平台,各政府部门、科研工作者可使用该服务器进行模块化的生物信息学分析[18]。GalaxyTrakr是美国食品安全与应用营养中心(Center for Food Safety and Applied Nutrition,CFSAN)建立在该服务器上的一个公共分析管道(pipeline)环境[19],其目的是快速识别和溯源食源性疾病的暴发,建立长期有效的食源性病原体监测系统,同时实现可跨实验室的数据共享与统一标准的分析处理步骤。GalaxyTrakr核心逻辑在于利用NCBI/EMBL/DDBJ等公共基因数据库中海量数据结合本部门测序数据作为总数据库,其日常监测的临床、食品及环境样本测序后,经模块化的序列拼接、质控、基因注释,进行数据库比对,即可鉴定出菌株的具体型别,同时可构建系统进化树进行溯源调查。该模式的最大优点为:1)其构建的生物信息分析模块是开源且可通过网络访问的;2)该模块为非生物信息学专家提供了标准的自动分析操作和可视化界面;3)数据可跨实验室共享,结果可复现;4)既可在线云平台分析也可部署到本地计算机,且分析流程可扩展。
2.2.2 美国PulseNet实验室监测体系
1996年,美国疾控中心联合国内多家实验室共同启动PulseNet计划,旨在建立一个统一的国家实验室监测系统,用于快速检测美国境内各病原体引起的暴发性疾病(食源性为主)[5-6]。过去几十年间该监测系统一直使用PFGE作为病原微生物检测的金标准。随着NGS技术的快速发展与测序成本的不断降低,2013年PulseNet开始使用WGS测序技术调查食源性疾病暴发事件。截至2018年,50多家实验室全部采用WGS技术进行常规监测,至此该技术成为新的金标准。
该实验室网络采用平均核苷酸一致性(Average Nucleotide Identity,ANI)方法鉴定病原类别;血清型、耐药/毒力基因识别等分析采用丹麦技术大学基因组流行病学中心的工具,所有工具都整合自软件BioNumerics中[5, 20-21]。该监测体系实际应用案例,例如2016年美国两州同时暴发同PFGE型别(JEGX01.002)的肠炎沙门氏菌(SE)感染[22],经全基因测序后进行系统发育分析(图2),发现两地暴发菌株相互独立单独成簇,实际并无关联性。
据相关研究估计[23-24],该系统每年的运营成本约为1000万~1500万美元,其在执行实时监测期间,预防了约270000例食源性疾病的发生,每年为该国节省超过5亿美元的医疗费用和经济损失。PulseNet成功运行的关键之一是所有成员都使用统一标准的分析方法,从而可以实现快速高效的数据共享和比较分析。
该系统因采用WGS方法,测序前需对样本中病原菌进行分离培养,因不同菌种分离培养方法及条件不同,增大了日常监测难度且延长了进度时间。另外,其仅适用于固定细菌种类的常规监测,难以发现监测种类以外的和未知的病原菌。因mNGS技术可直接从各类样本中回收、测序其所含全部遗传序列,无需进行培养,未来PulseNet或许会再次进行技术升级。
2.2.3 国家致病菌识别网
中国法定传染病报告系统在2004年全面升级改造后,建立了全国统一的疾病信息网络数据库,该数据库包含了庞大的临床诊断和基础流行病学信息,但并未关联到鉴定疾病最核心的病原体数据。中国疾控中心传染病预防控制所于2009年6月正式加入PulseNet International项目,成立PulseNet China,建立了中心、省、地市三级实验室网络[25]。2017年,传染病预防控制所依靠现有的PulseNet China实验室体系,建立了国家致病菌识别网(Chinese Pathogen Identification Net,China PIN),该监测网络旨在使用NGS测序技术进行病原鉴定及遗传信息分析,如耐药性、毒力基因、分子分型等,从而对具有流行风险的致病菌进行常规监测,及对已发生的疾病暴发事件进行基因组溯源调查[26]。
该系统具有以下优点:1)联动各级疾控部门;2)优于传统的单纯针对“率”的监测,可提供生物学证据;3)序列数据可共享,结果可重现;4)精确到菌株亚型,可进行精准暴发溯源;5)兼顾科研,提供其他基因组特征分析。但该系统当前仍处于建设阶段,且受实验室仪器、技术水平等影响,省、市级实验室仍以PFGE等传统湿实验为主,较少开展全基因组测序。另一明显限制为所监测病原种类及来源较为局限,目前仅疾控系统内部收集包含大肠埃希菌、副溶血弧菌、空肠弯曲菌、沙门氏菌等若干食源性致病菌。
2.2.4 Nextstrain开放社区项目
除政府部门组建的基于NGS测序的新型传染病监测预警平台,还有众多开放团体维护的监测平台,如Nextstrain,其为全球众多科研人员共同维护的病原进化监测开源平台[27-28]。该平台并不属于狭义的传染病监测预警,而是通过实时的系统发育分析更新全球范围致病病原的最新进化、变异情况,从而提示当前及潜在的公共卫生风险[28]。该项目关注登革热、埃博拉病毒、肠病毒-D68(EV-D68)、流感、猴痘、西尼罗病毒、寨卡病毒等致病源。新冠大流行期间,该平台以GISAD数据库测序数据为主,对新冠病毒(SARS-CoV-2)的流行及变异情况进行实时追踪,所有数据免费开放。
3 海关系统NGS传染病监测预警平台初步设想
3.1 整体架构
我国海关系统可利用现有的实验室体系建成1~2中心、多站点的传染病监测预警实验室网络,云平台服务器部署于中心实验室,全国各站点实验室可通过在线访问中心服务器或利用docker等容器技术将相同的标准分析流程安装至本地计算机进行分析,如病原菌的鉴定识别及系统发育树的构建等。中心实验室负责NGS数据存储、服务器及分析模块开发维护,站点实验室负责所属海关的常规监测及检测任务。
站点实验室技术人员无需具有生物信息学背景,只需在可视化页面通过简单点击操作即可完成一系列分析工作。前期阶段,当前现有的成熟简便的实验室检测手段可用于海关常规布控指令的监测采样等,NGS测序分析用于发现高风险个体或发生公共卫生事件后的一系列精准鉴定及溯源分析。
平台功能围绕以下几点为宜:1)病原菌鉴定;2)判断跨境传播事件;3)精准溯源,揭示传播动态;4)早期暴发预警;5)识别未知病原;6)评估口岸预防及控制的措施效果;7)其他科学研究。
经长期运行,该平台可积累海量关联详细流行病学信息的人、动植物、物品来源的病原基因组序列,不仅可为后期的相关分析提供精准翔实的本底数据,还可为未来可能的业务职能扩展提供客观条件。同时,该平台的丰富数据及经典的人群代表性可促进与各国际卫生组织、科研院所的合作交流及自身良性发展。
3.2 技术层设计
结合海关特点,技术分析建议采用以下4项原则:1)模块化、流程化、统一化;2)数据可跨实验室,结果可复现;3)关联流行病学信息;4)重视干实验,逐步淘汰湿实验。
第一项原则具体指当前各地实验室技术水平及检测方式不同,可能造成检测结果及解读的不统一及实验数据的“多源异构”特征,搭建统一的云操作平台,有利于上述问题的标准化。第二项原则并非指所有人均可获得实验数据,而是指实验数据具有统一的命名、分析和方案解读规则。举例说明,大肠埃希菌基于7个管家基因的MLST分型方案较为成熟且为业内共识。基于该分型方案得出的ST型别,其他实验室拿到原始数据也可得出相同ST型结果,且分析方法相同、解读相同,有利于样本分析的互联互通。第三项目原则为后期的流行病学及统计学分析提供基础。第四项原则明确了该平台立足未来的技术前沿性。
3.3 执行层现状及优化
当前,针对入境人员的卫生检疫仍以旅客主动申报、体温监测和医学巡查为主,医学巡查即现场关员采用目测等办法对入境人员进行观察判断。对于重点地区的入境人员,流程一般为检查有效的预防接种证书,体温检测及观察是否具有相关症状。对有疑似相关症状的人员进行流行病学调查并采集样本,然后进行单一或若干病原的快速检测。此类传统检疫、检测方式面对隐匿性强的疾病或潜伏期病人,具有一定的漏查漏检风险。但对重点人员利用传统实验方法检测所有法定传染病及高危性病原并不具备现实可操作性。
针对该情况,在不更改现行海关检疫制度的前提下,通过检测平台的升级,也可显著扩大传染病检测范围,减小漏检风险。利用本文设计的监测预警平台,可通过采集重点人员样本(粪便、血液等),不经过菌株分离培养直接进行宏基因组测序,所得测序数据可快速、精准地鉴定出样本所携带的几乎全部病原微生物种类。重点动植物及商品(如冷链食品)样本同样可进行相关分析。需要注意的是,短期之内在实际执行层面该技术全面铺开难度较大且费用较高,可在重点监测群体的检疫中作为现有模式的技术补充。
4 讨论
传统传染病监测预警多为收集本地区传染病发生发展的有关信息,在时间、空间、人群三个维度对发病率、病死率等一系列指标进行综合研判的系统[16-17]。随着互联网技术的快速发展,信息获得的便捷性极大提高,各国卫生机构对传染病暴发事件公布的透明化,使得传染病监测预警体系中信息收集渠道极大扩展,监测范围不仅仅局限于某一地区或国家[15]。但这种监测模式本质上仍是传统传染病监测预警的思路,未解决其本身固有的多种缺陷。
新型传染病往往流行一段时间后才能被传统监测平台识别,如2003年SARS流行及2019年起始的COVID-19大流行[29-32]。基于NGS测序的生物信息学分析则有能力在监测样本中识别未知病原,当前该模式已经成为传染病病原学监测、暴发调查和分子流行病学分析中的主流手段。新型的传染病识别监测网络多采用该技术并取得了极大的成功,如上文提到的美国PulseNet项目及正在建设阶段的国家致病菌识别网。虽然当前NGS测序费用及测序时间相较传统检测方式较高,但从一次测序即可得出众多实验结果的角度来看,其性价比更高且未来测序费用仍将大幅下降[2, 33]。
参考国内外众多监测体系,结合海关职责,建议在此契机下建立一套采用未来主流技术方向,以识别、防范高危的已知及未知病原输入为目的,同时兼顾输入后精准溯源的全国各口岸统一实验室监测平台。其既可承担口岸传染病常规监测预警,又可在出现与出入境关联的重大公共卫生事件后,立即进行病原精准鉴定、基因组溯源工作,以期为海关系统的防控政策及对外发声提供科学证据。在疫情防控期间,海关系统积累了相关检测技术手段及未来基因组测序设备储备,这也为该体系的组建提供了一定客观条件。
参考文献
[1] Mccombie W R, Mcpherson J D, Mardis E R. Next-Generation Sequencing Technologies[J]. Cold Spring Harb Perspect Med, 2019, 9(11): a036798. DOI: 10.1101/cshperspect.a036798
[2] Hu T, Chitnis N, Monos D, et al. Next-generation sequencing technologies: An overview[J]. Human Immunology, 2021, 82(11): 801-811.
[3] Yoshinaga Y, Daum C, He G, et al. Genome Sequencing[J]. Methods in Molecular Biology, 2018, 1775: 37-52.
[4] Li N, Cai Q, Miao Q, et al. High-Throughput Metagenomics for Identification of Pathogens in the Clinical Settings[J]. Small Methods, 2021, 5(1): 2000792. DOI: 10.1002/smtd.202000792
[5] Ribot E M, Freeman M, Hise K B, et al. PulseNet: Entering the Age of Next-Generation Sequencing[J]. Foodborne Pathogens and Disease, 2019, 16(7): 451-456.
[6] Tolar B, Joseph L A, Schroeder M N, et al. An Overview of PulseNet USA Databases[J]. Foodborne Pathogens and Disease, 2019, 16(7): 457-462.
[7] Pérez-Losada M, Arenas M, Castro-Nallar E. Microbial sequence typing in the genomic era[J]. Infection Genetics And Evolution, 2018, 63: 346-359.
[8] Kimura B. Will the emergence of core genome MLST end the role of in silico MLST?[J]. Food Microbiology, 2018, 75: 28-36.
[9] Li J, Lai S, Gao G F, et al. The emergence, genomic diversity and global spread of SARS-CoV-2[J]. Nature, 2021, 600(7889): 408-418.
[10] Lauring A S, Hodcroft E B. Genetic Variants of SARS-CoV-2-What Do They Mean?[J]. Jama, 2021, 325(6): 529-531.
[11] Lucey M, Macori G, Mullane N, et al. Whole-genome Sequencing to Track Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Transmission in Nosocomial Outbreaks[J]. Clinical Infectious Diseases, 2021, 72(11): 727-735.
[12] Nelson M I. Tracking the UK SARS-CoV-2 outbreak[J]. Science, 2021, 371(6530): 680-681.
[13] Woodall J, Calisher C H. ProMED-mail: background and purpose[J]. Emerging Infectious Diseases, 2001, 7(3): 563. DOI: 10.3201/eid0707.017736
[14] Rolland C, Lazarus C, Giese C, et al. Early Detection of Public Health Emergencies of International Concern through Undiagnosed Disease Reports in ProMED-Mail[J]. Emerging Infectious Diseases, 2020, 26(2): 336-339.
[15] Carrion M, Madoff L C. ProMED-mail: 22 years of digital surveillance of emerging infectious diseases[J]. International Health, 2017, 9(3): 177-183.
[16] Wang L, Wang Y, Jin S, et al. Emergence and control of infectious diseases in China[J]. Lancet, 2008, 372(9649): 1598-1605.
[17] 杨维中, 兰亚佳, 李中杰, 等. 国家传染病自动预警系统的设计与应用[J]. 中华流行病学杂志, 2010, 31(11): 1240-1244.
[18] Jalili V, Afgan E, Gu Q, et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2020 update[J]. Nucleic Acids, 2020, 48(1): 395-402.
[19] Gangiredla J, Rand H, Benisatto D, et al. GalaxyTrakr: a distributed analysis tool for public health whole genome sequence data accessible to non-bioinformaticians[J]. BMC Genomics, 2021, 22(1): 114. DOI: 10.1186/s12864-021-07405-8
[20] Bortolaia V, Kaas R S, Ruppe E, et al. ResFinder 4.0 for predictions of phenotypes from genotypes[J]. Journal of Antimicrobial Chemotherapy, 2020, 75(12): 3491-3500.
[21] Clausen P, Aarestrup F M, Lund O. Rapid and precise alignment of raw reads against redundant databases with KMA[J]. BMC Bioinformatics, 2018, 19(1): 307. DOI: 10.1186/s12859-018-2336-6.
[22] Kubota K A, Wolfgang W J, Baker D J, et al. PulseNet and the Changing Paradigm of Laboratory-Based Surveillance for Foodborne Diseases[J]. Public Health Reports, 2019, 134(2): 22-28.
[23] Scharff R L, Besser J, Sharp D J, et al. An Economic Evaluation of PulseNet: A Network for Foodborne Disease Surveillance[J]. American Journal of Preventive Medicine, 2016, 50(5): 66-73.
[24] Ribot E M, Hise K B. Future challenges for tracking foodborne diseases: PulseNet, a 20-year-old US surveillance system for foodborne diseases, is expanding both globally and technologically[J]. EMBO Reports, 2016, 17(11): 1499-1505.
[25] Xu J. PulseNet China[J]. Emerging Infectious Diseases, 2012, 1(10): e29. DOI: 10.1038/emi.2012.30
[26] Cui Z, Zhou H, Meng S, et al. Chinese Pathogen Identification Net: A Laboratory Network for Surveillance and Response of Bacterial Infectious Diseases[J]. China CDC Weekly, 2022, 4(12): 235-237.
[27] Hadfield J, Megill C, Bell S M, et al. Nextstrain: real-time tracking of pathogen evolution[J]. Bioinformatics, 2018, 34(23): 4121-4123.
[28] Morel B, Barbera P, Czech L, et al. Phylogenetic Analysis of SARS-CoV-2 Data Is Difficult[J]. Molecular Biology And Evolution, 2021, 38(5): 1777-1791.
[29] Stadler K, Masignani V, Eickmann M, et al. SARS--beginning to understand a new virus[J]. Nature Reviews Microbiology, 2003, 1(3): 209-218.
[30] Zhong N S, Zheng B J, Li Y M, et al. Epidemiology and cause of severe acute respiratory syndrome (SARS) in Guangdong, the People’s Republic of China, in February, 2003[J]. Lancet, 2003, 362(9393): 1353-1358.
[31] Guo Y R, Cao Q D, Hong Z S, et al. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak-an update on the status[J]. Military Medical Research, 2020, 7(1): 11.
[32] Xu S, Li Y. Beware of the second wave of COVID-19[J]. Lancet, 2020, 395(10233): 1321-1322.
[33] Mitchell S L, Simner P J. Next-Generation Sequencing in Clinical Microbiology: Are We There Yet?[J]. Clinics In Laboratory Medicine, 2019, 39(3): 405-418.
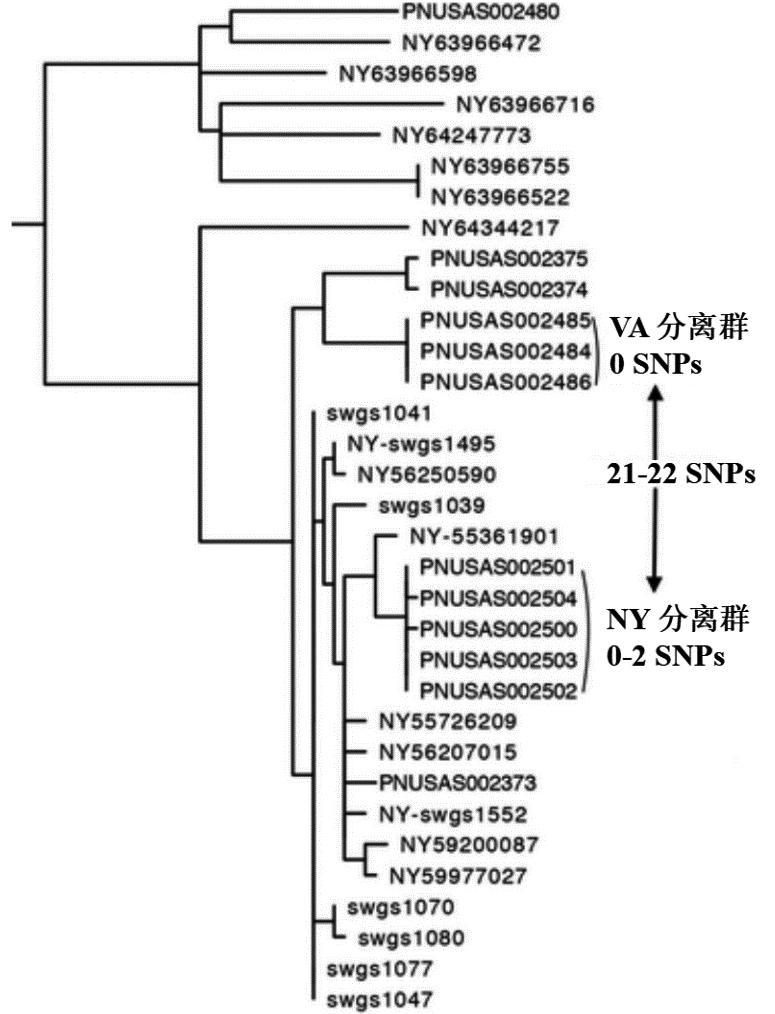
单核苷酸多态性 (Single Nucleotide Polymorphisms, SNP) 即基因组水平上由单个核苷酸的变异所引起的DNA序列多态性.
图2 美国纽约州和弗吉尼亚州中肠炎沙门氏菌暴发案例的系统发育树[22]
Fig.2 A phylogenetic tree of Salmonella outbreak cases from New York and Virginia, USA
