





CopyRight 2009-2020 © All Rights Reserved.版权所有: 中国海关未经授权禁止复制或建立镜像

在线固相萃取-液相色谱-高分辨质谱法快速测定牛奶中10种利尿剂残留量
作者:黄雪静 李颖 郄俊青 王敬 张婧雯 魏欣欣 张海超
黄雪静 李颖 郄俊青 王敬 张婧雯 魏欣欣 张海超
摘 要 本研究基于在线固相萃取-液相色谱-四极杆/静电场轨道阱高分辨质谱建立了同时测定牛奶中10种利尿剂的测定方法。试样采用乙腈提取,提取液经在线固相萃取柱净化,待测物在CAPCELL PAK ADME(150 mm×2.1 mm,5 µm)色谱柱上用乙腈-水进行梯度洗脱分离。同时,采用加热电喷雾电离源(Heat Electric Spray Ion Source,HESI)以正负离子全扫描/数据依赖二级扫描模式进行分析,外标法定量。结果显示,10种利尿剂在各自线性范围内线性关系良好,相关系数(r)大于0.9978,检出限(LODs)为1~3 µg/kg,定量限(LOQs)为3~10 µg/kg,平均回收率在73.6%~104.2%之间,相对标准偏差(RSDs)在3.6%~8.1%之间。该方法操作简单、灵敏度高、准确性好,可为相关检测人员提供更高效的解决方案。
关键词 在线固相萃取;液相色谱;静电场轨道阱高分辨质谱;利尿剂;牛奶
Rapid Determination of 10 Diuretics in Milk by
on-Line Solid Phase Extraction Coupled with Liquid Chromatography-High Resolution Mass Spectrometry
HUANG Xue-Jing1 LI Ying1 QIE Jun-Qing1 WANG Jing1
ZHANG Jing-Wen1 WEI Xin-Xin1 ZHANG Hai-Chao1*
Abstract A novel method was developed for the rapid determination of 10 diuretics in milk by on-line solid phase extraction coupled with liquid chromatography-quadrupole/electrostatic field orbitrap high-resolution mass spectrometry (Online-LC-Q/Orbitrap HRMS). The samples were extracted with acetonitrile, then purified by an on-line solid phase extraction column, and subsequently separated on an CAPCELL PAK ADME (150 mm×2.1 mm, 5 µm) column using a gradient elution with acetonitrile-water as the mobile phase. The diuretics were analyzed employing a Heat Electric Spray Ion source (HESI) in both positive and negative ion Full Scan/ddMS2 modes, and quantification was performed using the external standard method. The results demonstrated excellent linearity within the tested concentration ranges, with correlation coefficients (r) greater than 0.9978. The limits of detection (LODs) ranged from 1-3 µg/kg, and the limits of quantitation (LOQs) from 3-10 µg/kg. The average recoveries for the 10 diuretics were between 73.6% and 104.2%, with relative standard deviations (RSDs) of 3.6%-8.1%. This method is straightforward to perform, highly sensitive, and accurate, offering a more efficient solution for detection personnel.
Keywords on-line solid phase extraction; liquid chromatography; electrostatic field obitrap high-resolution mass spectrometry; diuretics; milk
基金项目:海关总署科研项目(2019HK112)
第一作者:黄雪静(1990—),女,汉族,河北隆尧人,本科,工程师,主要从事食品残留分析工作,E-mail: 1213143644@qq.com
通信作者:张海超(1986—),女,汉族,河北满城人,硕士,工程师,主要从事食品安全分析工作,E-mail: haichao0602@163.com
1. 石家庄海关技术中心 石家庄 050051
1. Shijiazhuang Customs Technology Center, Shijiazhuang 050051
利尿剂是一类促进体内水分和电解质排出的药物,在临床上可用来治疗心、肝、肾等各类水肿,在畜禽养殖中常被用作治疗奶牛乳房炎[1-2]。目前关于利尿剂残留限量的规定较少,仅在GB 31650—2019《食品安全国家标准 食品中兽药最大残留限量》中规定允许将氢氯噻嗪用于牛的养殖过程[3]。因此,建立一种操作简单、快速灵敏测定牛奶中利尿剂的分析方法对于牛奶品质的监管、维护人们身体健康都具有重要意义。
关于利尿剂的检测方法主要有液相色谱法[4-5]、气相色谱-质谱法[6-7]、液相色谱-串联质谱法[8-11]。液相色谱-串联质谱法因具有较好的灵敏度且前处理过程相对简便而在动物源性食品利尿剂的测定中得到广泛应用。但随着高分辨质谱的普及,也有研究报道将液相色谱-四极杆/静电场轨道阱质谱用于该类药物的测定[12-13],提高了定性的准确性。Ji Hyun Lee等[12]对膳食补充剂中23种利尿剂进行了筛查及裂解原理的阐述。张丽华等[13]采用离线净化和浓缩的方式对动物源性食品中25种利尿剂进行了分析,检出限为5~10 μg/kg。本研究选择了日常监测的10种利尿剂,开发了在线固相萃取技术对样品进行净化浓缩的手段,旨在克服传统固相萃取[4-6,8-10,13]和QuEChERS[11,14]等离线净化方式中存在的费时费力、重现性差等问题。
1 实验部分
1.1 仪器与试剂
在线净化液相色谱-四极杆/静电场轨道阱高分辨质谱仪Q-Orbitrap(美国ThermoFisher公司);Cyclone-P聚合物在线净化柱,0.5 mm×50 mm,60 μm(美国ThermoFisher公司); CAPCELL PAK ADME色谱柱,150 mm×2.1 mm,5 μm(日本SHISEIDO公司);离心机,Sigma 3K-15型,(德国Sigma公司);高速均质器,PT2100型(瑞士KINEMATICA公司);涡旋混合器(美国Scientific Industries);超纯水系统,Advantage A10+ELIX5型(美国Millipore公司)。
乙腈、甲醇、丙酮、异丙醇、甲酸、乙酸、乙酸铵为色谱纯(美国Thermo Fisher公司);无水硫酸钠、氯化钠为分析纯(天津大茂公司);水为Milli-Q高纯水。
标准品:乙酰唑胺、坎利酮、氯噻酮、呋塞米、螺内酯、苄氟噻嗪、氯噻嗪、氢氯噻嗪、氨苯蝶啶、精磺胺(德国Dr. Ehrenstorfer公司),纯度大于97%。
1.2 混合标准溶液的配制
分别称取10种利尿剂标准品10 mg于10 mL棕色容量瓶中,用甲醇溶解、定容,得到浓度为1 mg/mL的单标储备液,于4℃下避光保存。根据需要,准确移取10种利尿剂标准储备液,用甲醇稀释成10 μg/mL混合标准工作液,待用。
1.3 样品前处理
称取10 g(精确到0.01 g)均匀试样,置于50 mL离心管中,加入20 mL乙腈,4 g氯化钠迅速摇匀,用均质器以10000 r/min 均质1 min后,在5000 r/min下离心5 min。移取1 mL提取液加入1 mL 0.1%甲酸水溶液涡旋混匀,过0.22 μm的有机尼龙滤膜,进样测定。
1.4 在线固相萃取与液相分离条件
在线固相萃取色谱条件(Online SPE):流动相A为10 mmol/L乙酸铵水溶液(含0.02%乙酸)、B为乙腈、C为丙酮+乙腈+异丙醇(1∶1∶1,V∶V∶V),进样量为50 μL。液相分离色谱条件:流动相A为水,流动相B为乙腈,其在线净化梯度洗脱程序见表1。整个过程采用2个六通阀切换流路,1.0~3.0 min二元泵将富集在固相萃取柱上的利尿剂目标物洗脱至色谱上柱开始进行分离检测。在线净化程序中每步的变化采用瞬变模式,液相分离每步的变化除最后一步采用瞬变模式外其他步骤间改变采用渐变模式。整个运行时间为26 min。
1.5 质谱条件
离子源:加热电喷雾离子源(Heat Electric Spray Ion Source,HESI);喷雾电压:3200 V;离子传输管温度:350℃ ;辅助加热气温度:320℃ :扫描模式:一级母离子全扫描和数据依赖的二级子离子扫描模式(Full Scan/ddMS2);扫描范围(m /z):100~500,一级质谱扫描分辨率为70000 FWHM,二级质谱扫描分辨率为17500 FWHM;归一化碰撞能量为NCE为40 eV (±50% )。分析物质谱信息见表2。
2 结果与讨论
2.1 前处理条件的优化
10种利尿剂大多同时含有酸性和碱性基团,性质差异较大。实验对比了乙酸乙酯和乙腈的提取效果,二者的回收率无明显差异。考虑到与后续处理方法的兼容性,所以本实验选择乙腈作为提取溶剂。样品定容液为在线固相萃取的上样溶液,影响着化合物在净化柱上的保留情况和加标样品的回收率。因此,实验比较了乙腈和0.1%甲酸水不同比例混合溶液(乙腈和0.1%甲酸水比例分别为9∶1、7∶3、1∶1、3∶7、1∶9)作为定容液时目标物在净化柱上的保留情况。当乙腈含量为90%时,化合物乙酰唑胺、氯噻嗪、氢氯噻嗪和精磺胺化合物不保留,其他化合物部分保留。随着乙腈比例的减小,各化合物在净化柱上的保留行为逐步增强,当乙腈降至比例为50%时,各化合物在净化柱的保留部分无明显变化。虽然随着乙腈比例的降低,各化合物保留增强,但是回收率降低。所以综合选择乙腈-0.1%甲酸水(1∶1,V∶V)作为定容液。
2.2 色谱条件的优化
本研究采用ADME色谱柱进行分析,通过对流动相比例的不断优化,采用表1梯度洗脱条件可实现对化合物的保留和分离。实验为保证各化合物最佳的分析效果,对流动相水-乙腈、0.1%甲酸水-乙腈、5 mmol/L乙酸铵-乙腈、水-甲醇进行了比较,同时对流动相初始比例和梯度洗脱条件进行了优化,10 ng/mL的各化合物响应如图1所示,可以看出采用水-乙腈对各化合物响应值最高。
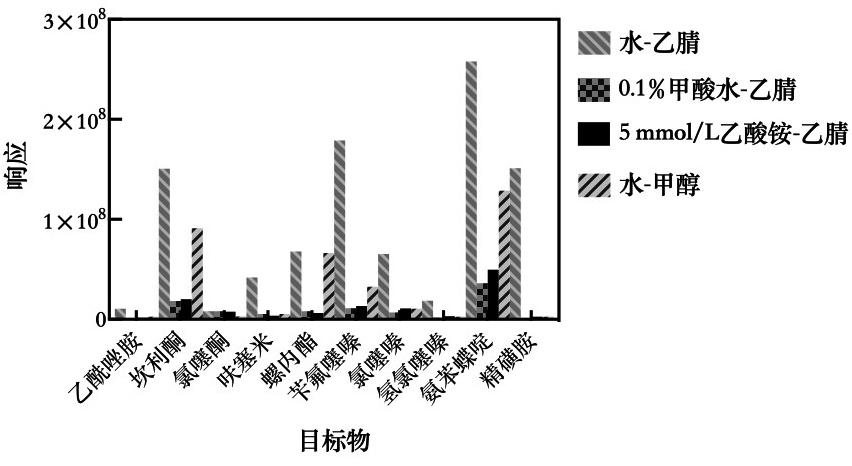
图1 流动相对10种利尿剂灵敏度的影响(10 ng/mL)
Fig.1 Effects of mobile phase on the sensitivity of 10 diuretics (10 ng/mL)
2.3 质谱条件的选择和优化
本实验采用全扫描和正、负离子切换模式进行测定。通过一级质谱的精确质量数进行定性定量,并设置自动触发二级模式提高定性的准确性。在一级扫描质谱图中发现这10种化合物在ESI-和ESI+模式下响应各不相同,坎利酮和氨苯蝶啶只在正离子模式有响应,为[M+H]+峰。螺内酯在高温源内不稳定,容易脱去一分子乙酰基硫基后生成质荷比为341.2111的正离子峰,与坎利酮母离子相同。乙酰唑胺、氯噻酮和氯噻嗪在正负模式下均有响应,但负模式响应值较高,选择[M-H]-峰作为母离子。其他4种化合物呋塞米、苄氟噻嗪、氢氯噻嗪和精磺胺仅在负模式下灵敏度较高,母离子均为[M-H]-峰。故本方法采用正负同时全扫描模式和数据依赖扫描模式进行化合物的测定,采用较高的分辨率(70000 FWHM)在一定的质量数范围做一级质谱全扫描,以保证目标化合物具有更好的质量准确度。在扫描范围内母离子的强度达到设定阈值(1×105)时自动触发二级子离子质谱扫描,采用40 eV (±50% )的碰撞能量可得到二级质谱信息,选用丰度较高的碎片离子作为特征离子,最终建立的质谱条件见表2。10种利尿剂的选择离子流图如图2所示。
2.4 基质效应评价
基质效应(Matrix Effect,ME)指测定目标物以外的其余组分对测定结果的影响。实验通过比较基质标准曲线和溶剂标准曲线的斜率比来判断基质效应的强弱。计算公式如下:ME =基质标准曲线斜率/溶剂标准曲线斜率[16]。当比值在0.8~1.2之间时基质效应最弱,比值大于1.2或小于0.8时基质效应显著。如图3显示,氯噻酮、呋塞米、氯噻嗪和精磺胺比值在0.23~0.72之间,表现出较强的基质抑制效应;其余化合物的基质效应均较弱,在0.82~1.18之间。由于各化合物在基质中离子化效率表现不同,为消除基质效应对分析结果的影响,本方法采用基质标准溶液对结果进行校正,使目标物在标准工作溶液和样品溶液中均具有相似的离子化环境,确保方法定性和定量的准确性。
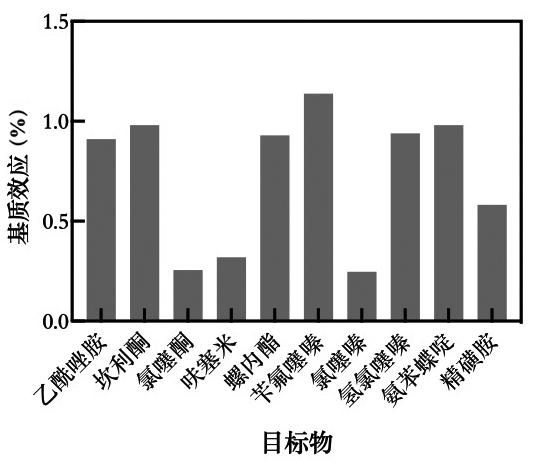
图3 10种利尿剂的基质效应
Fig.3 Matrix effects of 10 diuretics
2.5 线性范围、检出限和定量限
根据分析物的灵敏度,选用牛奶的空白基质溶液配制了基质匹配标准溶液,以待测物峰面积Y为纵坐标,对应质量浓度X为横坐标,绘制标准曲线。结果表明,10种利尿剂在一定的质量浓度范围内均呈良好线性关系,相关系数(r)在0.9978~0.9997之间,采用逐级稀释的方式,并结合信噪比S/N>3以及S/N>10的要求,确定方法的检出限(LODs)和定量限(LOQs)。结果表明,10种利尿剂的检出限为1~3 μg/kg,定量限为3~10 μg/kg。线性方程、线性范围、相关系数、检出限和定量限见表3。
2.6 方法的回收率、精密度
取已制备均匀的空白样品,准确加入10种利尿剂的混合标准溶液,分别添加浓度为3 μg、10 μg、50 μg /kg的样品各6份,按上述优化后的方法进行分析。采用各空白基质溶液配制系列标准溶液,外标法进行定量分析,计算平均回收率和相对标准偏差(RSDs)。10种利尿剂的回收率均在73.6%~ 104.2%之间,RSDs在3.6%~8.1%之间,能够满足牛奶中利尿剂的定性和定量的要求。牛奶的加标回收率和相对标准偏差见表4。
2.7 与标准方法比较
为进一步验证该方法的有效性,本实验采用与标准方法比对的方式对加标为25 μg/kg氢氯噻嗪的样品进行检测,平行测定3次,本方法测定值为23.1 μg/kg、24.6 μg/kg和23.5 μg/kg,GB 31658.25—2022《食品安全国家标准 动物性食品中10种利尿剂残留量的测定 液相色谱-串联质谱法》[15]的测定值分别为24.6 μg/kg、24.0 μg/kg、22.9 μg/kg,采用t检验对两组数据进行统计分析,t为0.38,小于t(0.05,5)临界值2.78,两组数据不具有明显性差异,可见本方法的准确度良好。该加标样品的提取离子图及质谱如图4所示。
3 结论
本文基于在线固相萃取净化技术,建立了牛奶中10种利尿剂的四极杆/静电场轨道阱高分辨质谱检测方法。样品采用乙腈提取经0.1%甲酸水稀释后直接进样测定。与常规方法相比,直接进样在线固相萃取富集净化的方法大大简化了前处理过程,缩短了前处理时间。该方法操作简便,有机试剂使用量少,自动化程度高,可有效提高大批量样品的检测精度和检测效率,适用于牛奶中10种利尿剂的快速检测,有助于企业降低检测成本,能够为监管部门执法提供技术支撑。
参考文献
[1] 李文辉, 刘佳, 孙志文, 等. 动物源性产品中10种利尿剂残留的超高效液相色谱-串联质谱检测方法的研究[J]. 畜牧与兽医, 2022, 54(3): 53-58.
[2] 桂委. 利尿剂的临床应用及不良反应[J]. 医药世界, 2009, 11(4): 52.
[3] GB 31650—2019 食品安全国家标准 食品中兽药最大残留限量[S]. 北京: 中国标准出版社, 2019.
[4] Liu M, Lv B Q, Jiang H L, etal. Determination of diuretics in human urine using HPLC coupled with magnetic solid phase extraction based on a metal-organic framework[J]. Biomedical Chromatography, 2020, 34(9): 4876-4906.
[5] 秦旸, 朱绍棠, 王超, 等. 十三种利尿剂的高效液相色谱测定方法[J]. 分析测试学报, 2003, 22(1): 41-44.
[6] 赵海香, 郭振福, 汪丽萍, 等.气相色谱-质谱法测定茶饮料中6种利尿剂[J]. 理化检验(化学分册), 2010, 46(7): 820-828.
[7] 王培龙, 范理, 宋荣, 等. 气相色谱-质谱法确证分析饲料中6种利尿剂的研究[J]. 分析试验室,2009, 28(12): 31-34.
[8] 任晓伟, 范力欣, 何亮娜, 等. 超高效液相色谱-串联质谱法测定动物源性食品中的13种利尿剂[J]. 食品科学, 2023, 44(14): 360-367.
[9] 冯月超, 王建凤, 乔祎娜, 刘佳, 刘艳. 基于滤过型净化的液相色谱-串联质谱法测定畜禽肉中89种兽药残留[J]. 分析试验室, 网络首发.
[10] Yan Y H, Lian K Q, Zhang H C, etal. Doping-control analysis of 14 diuretics in animal-derived foods using ultra-high-performance liquid chromatography-tandem mass spectrometry[J]. Microchemical Journal, 2022, 174, 106948-106957.
[11] Chen D, Xu Q, Lu Y P, etal. The QuEChERS method coupled with high-performance liquid chromatography-tandem mass spectrometry for the determination of diuretics in animal-derived foods[J]. Journal of Food Composition and Analysis, 2021, 101, 103965-103972.
[12] Lee J H, Yang Y J, Min A Y, etal. Screening and elucidation of fragmentations of 23 diuretics in dietary supplements using UHPLC-Q-Orbitrap[J]. Science & Justice, 2021, 61(5): 451-458.
[13] 张丽华, 刘琪, 李星, 等. 超高效液相色谱-四极杆/静电场轨道阱高分辨质谱测定动物源食品中25种利尿剂[J]. 分析测试学报, 2023, 42(10): 1335-1342.
[14] 梁杨, 王小玲, 郭礼强. 基于QuEChERS提取的HPLC-MS/MS 法检测人血浆中6种常用利尿剂[J]. 药物分析杂志, 2019, 39(9): 1590-1596.
[15] GB 31658.25—2022 食品安全国家标准 动物性食品中10种利尿剂残留量的测定 液相色谱-串联质谱法[S]. 北京: 中国标准出版社, 2022.
[16]向平,沈敏,卓先义.液相色谱-质谱分析中的基质效应[J].分析测试学报, 2009, 28(6): 753-756.
表1 在线净化及色谱分离的梯度洗脱程序
Table 1 Gradient elution procedures of on-line cleanup and chromatographic separation
步骤 | 开始时间 (min) | 结束时间 (min) | 上样泵 | 洗脱泵 | ||||||
流速 (mL·min-1) | A (%) | B (%) | C (%) | 流速 (mL·min-1) | A (%) | B (%) | ||||
1 | 0.00 | 1.00 | 1.00 | 100 | — | — | 0.3 | 95 | 5 | |
2 | 1.00 | 2.00 | 0.10 | 60 | 40 | — | 0.3 | 95 | 5 | |
3 | 3.00 | 6.00 | 1.00 | — | — | 100 | 0.3 | 60 | 40 | |
4 | 9.00 | 9.50 | 1.00 | — | 100 | — | 0.3 | 45 | 55 | |
5 | 18.50 | 2.00 | 1.00 | — | 100 | — | 0.3 | 40 | 60 | |
6 | 20.50 | 1.50 | 1.00 | — | — | 100 | 0.3 | 10 | 90 | |
7 | 22.00 | 2.00 | 1.00 | 60 | 40 | — | 0.3 | 10 | 90 | |
8 | 24.00 | 2.00 | 1.00 | 100 | — | — | 0.3 | 95 | 5 | |
注: “—”表示该流路不运行
表2 10种利尿剂的名称、分子式及部分质谱参数
Table 2 Names, formulae and some MS parameters of 10 diuretics
名称 | 分子式 | 保留时间 (min) | 离子化模式 | 母离子 * ( m/z ) | 碎片离子 ( m/z ) | |
1 | 2 | |||||
乙酰唑胺 | C 4 H 6 N 4 O 3 S 2 | 7.90 | [M-H] - | 220.9809 | 83.0236 | 57.9741 |
坎利酮 | C 22 H 28 O 3 | 19.40 | [M+H] + | 341.2111 | 107.0859 | 187.1117 |
氯噻酮 | C 14 H 11 ClN 2 O 4 S | 11.06 | [M-H] - | 337.0055 | 146.0237 | 189.9728 |
呋塞米 | C 12 H 11 ClN 2 O 5 S | 8.16 | [M-H] - | 329.0004 | 204.9841 | 285.0114 |
螺内酯 | C 24 H 32 O 4 S | 20.07 | [M+H] + | 341.2111 | 107.0859 | 187.1117 |
苄氟噻嗪 | C 15 H 14 F 3 N 3 O 4 S 2 | 16.37 | [M-H] - | 420.0305 | 327.9688 | 289.0460 |
氯噻嗪 | C 7 H 6 ClN 3 O 4 S 2 | 8.53 | [M-H] - | 293.9415 | 213.9610 | 175.6928 |
氢氯噻嗪 | C 7 H 8 ClN 3 O 4 S 2 | 9.34 | [M-H] - | 295.9572 | 268.9463 | 204.9839 |
氨苯蝶啶 | C 12 H 11 N 7 | 14.65 | [M+H] + | 254.1149 | 237.0838 | 196.1689 |
精磺胺 | C 16 H 8 ClN 3 O 4 S 2 | 8.57 | [M-H] - | 283.9572 | 136.0508 | 169.0068 |
注: “*”表示定量离子
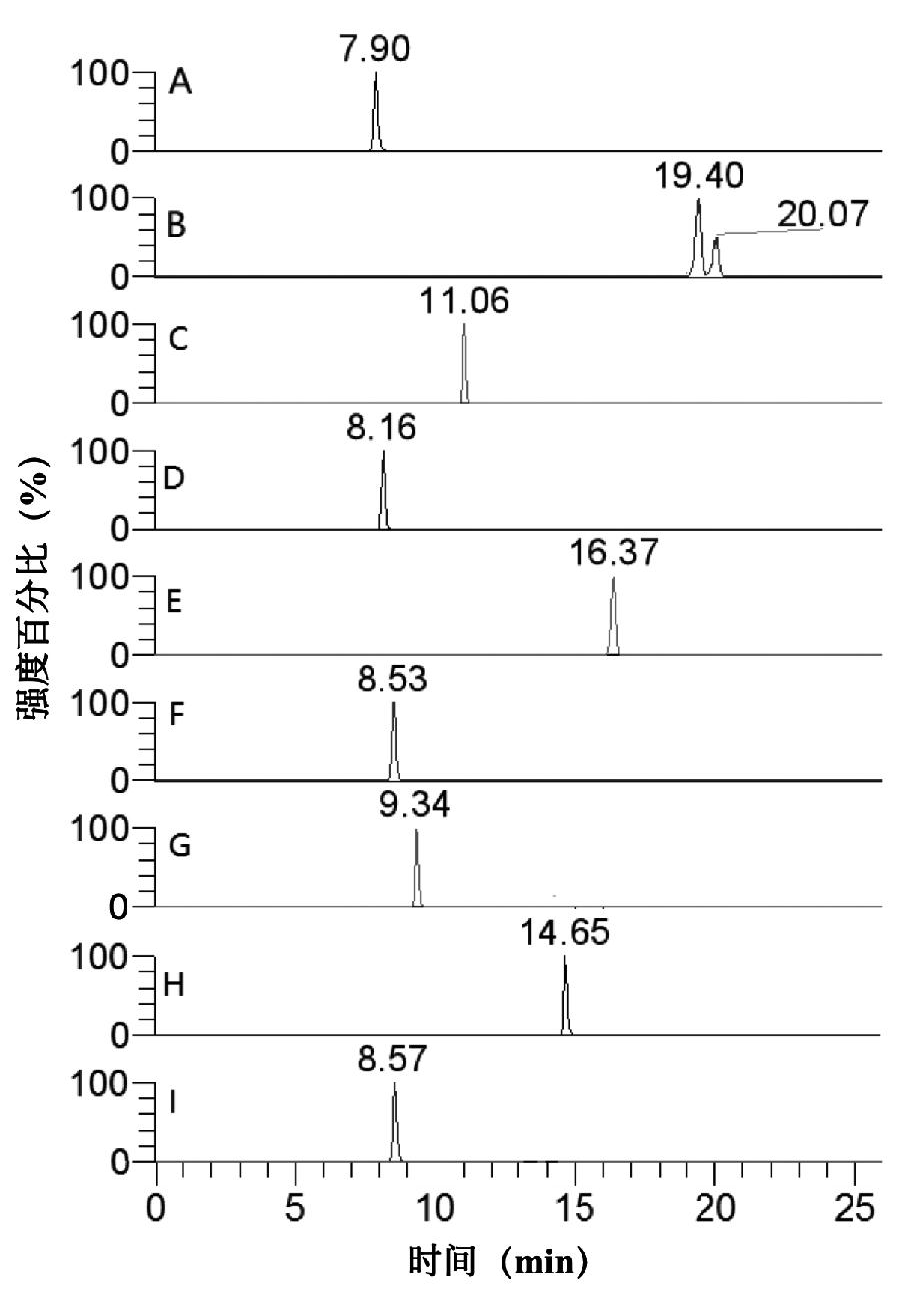
A: 乙酰唑胺; B: 坎利酮 (19.40 min) 和螺内酯 (20.07 min); C: 氯噻酮; D: 呋塞米; E:苄氟噻嗪; F: 氯噻嗪; G:氢氯噻嗪; H: 氨苯蝶啶; I: 精磺胺
图2 10种利尿剂的选择离子流图(10 ng/mL)
Fig. 2 XIC chromatograms of 10 diuretics (10 ng/mL)
表3 10种利尿剂的线性范围、相关系数、检出限和定量限
Table 3 Linear ranges, correlation coefficients (r), LODs and LOQs of 10 diuretics
化合物 | 线性方程 | 线性范围 (ng/mL) | 相关系数 (r) | LODs (μg/kg) | LOQs (μg/kg) |
乙酰唑胺 | Y = 313198X + 42406.2 | 0.5~50 | 0.9986 | 3 | 10 |
坎利酮 | Y = 3237080X + 2452510 | 0.2~50 | 0.9990 | 1 | 3 |
氯噻酮 | Y = 1375370X + 2220380 | 0.5~50 | 0.9978 | 3 | 10 |
呋塞米 | Y = 681204X + 157692 | 0.5~50 | 0.9986 | 2 | 6 |
螺内酯 | Y = 852068X + 294331 | 0.5~50 | 0.9995 | 2 | 6 |
苄氟噻嗪 | Y = 854656X + 1503280 | 0.2~50 | 0.9991 | 1 | 3 |
氯噻嗪 | Y = 1375370X + 2220380 | 0.2~50 | 0.9981 | 1 | 3 |
氢氯噻嗪 | Y = 682237X + 375627 | 0.5~50 | 0.9994 | 2 | 6 |
氨苯蝶啶 | Y = 21882500X-955960 | 0.2~50 | 0.9985 | 1 | 3 |
精磺胺 | Y = 583269X + 44215.1 | 0.5~50 | 0.9997 | 3 | 10 |
表4 方法的加标回收率及相对标准偏差(n = 6)
Table 4 Recoveries and RSDs of the method (n = 6)
化合物 | 添加 3 μg/kg | 添加 10 μg/kg | 添加 50 μg/kg | |||||
回收率 (%) | 相对标准偏差 (%) | 回收率 (%) | 相对标准偏差 (%) | 回收率 (%) | 相对标准偏差 (%) | |||
乙酰唑胺 | 82.0 | 7.8 | 85.1 | 6.0 | 90.9 | 6.6 | ||
坎利酮 | 78.3 | 7.9 | 86.8 | 7.1 | 91.2 | 4.8 | ||
氯噻酮 | 80.9 | 5.8 | 80.2 | 5.6 | 92.0 | 5.3 | ||
呋塞米 | 77.8 | 8.1 | 78.9 | 6.5 | 87.4 | 3.9 | ||
螺内酯 | 73.6 | 6.3 | 85.7 | 6.4 | 86.0 | 3.6 | ||
苄氟噻嗪 | 86.1 | 6.9 | 87.2 | 4.8 | 85.4 | 5.4 | ||
氯噻嗪 | 82.4 | 7.0 | 88.3 | 4.9 | 89.4 | 5.3 | ||
氢氯噻嗪 | 81.9 | 7.1 | 79.6 | 5.4 | 88.9 | 4.8 | ||
氨苯蝶啶 | 85.3 | 4.9 | 85.4 | 6.2 | 104.2 | 6.1 | ||
精磺胺 | 76.2 | 7.8 | 79.3 | 6.8 | 93.8 | 4.2 | ||
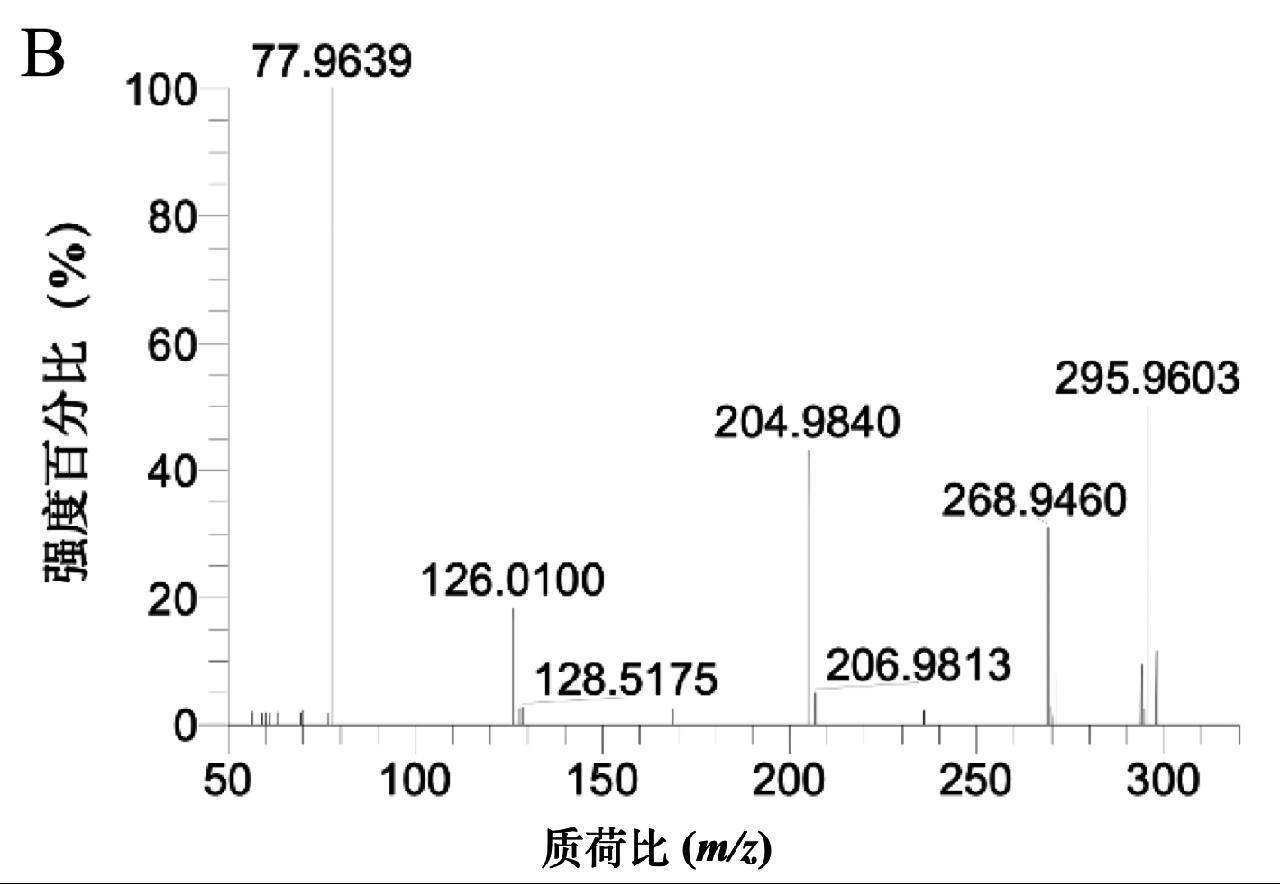
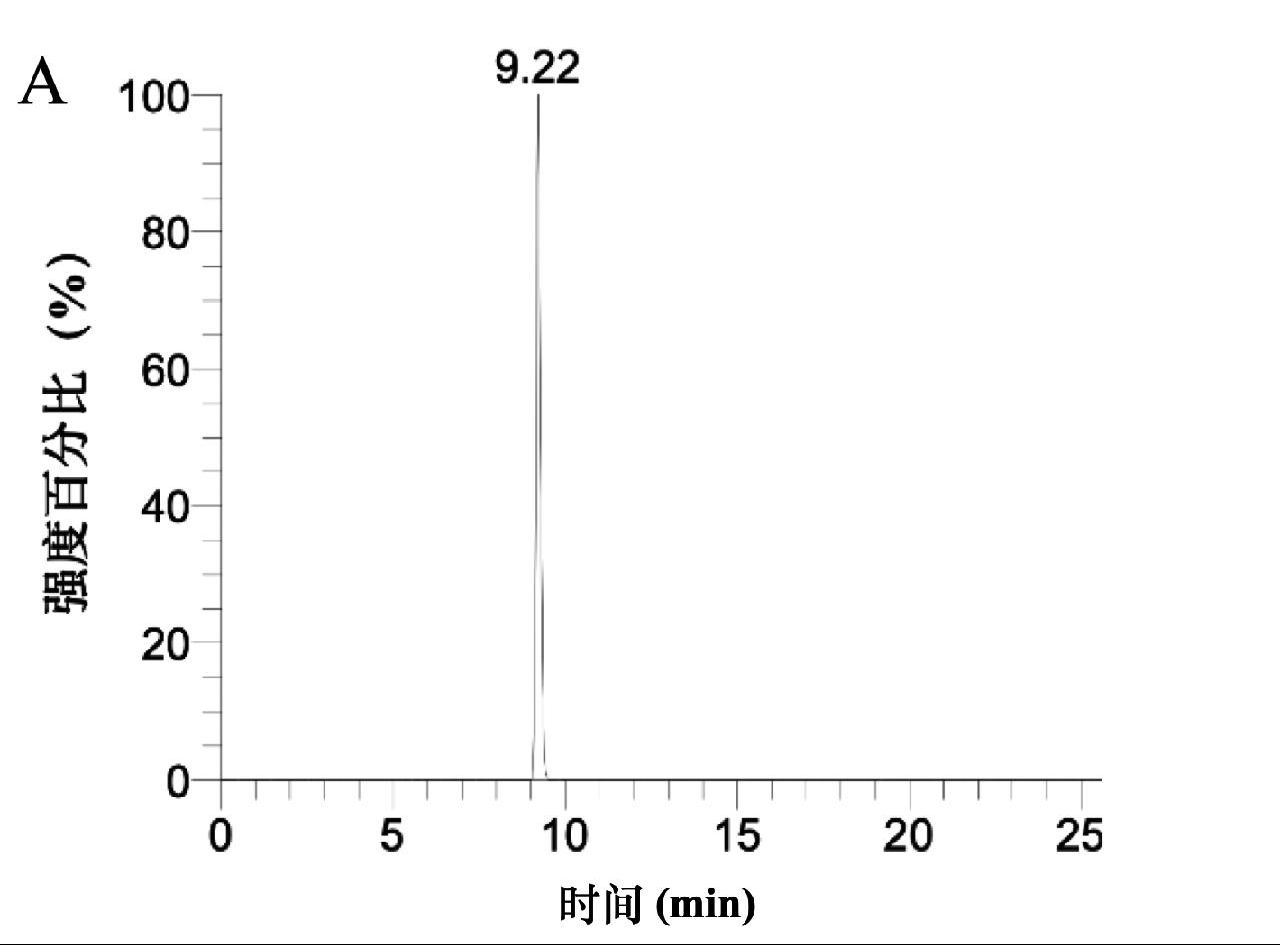
图4 添加样品(25 μg/kg)中氢氯噻嗪的提取离子色谱图(A)及质谱图(B)
Fig.4 Extracted ion chromatograms (A) and mass spectra of hydrochlorothiazide (B) in a spiked sample (25 μg/kg)
