





CopyRight 2009-2020 © All Rights Reserved.版权所有: 中国海关未经授权禁止复制或建立镜像

超高效液相色谱-三重四极杆-线性离子阱质谱快速检测动物源性食品中3种胃动力药物残留
作者:张宪臣 胡仪光 齐鹤鸣 李 勇 梁娜娜
张宪臣 胡仪光 齐鹤鸣 李 勇 梁娜娜
摘 要 本研究建立了超高效液相色谱-三重四极杆-线性离子阱质谱法快速检测畜肉、鱼肉和动物内脏等动物源性食品中3种胃动力药物(多潘立酮、莫沙必利和西沙必利)的方法。样品经乙酸乙酯提取、浓缩、复溶,采用20 mmol/L乙酸铵水溶液和甲醇溶液梯度洗脱,Agilent poroshell C18色谱柱(100 mm×3.0 mm,2.7 μm)进行分离;质谱采用电喷雾离子源(ESI)正离子检测,多重反应检测(MRM)模式下进行内标法定量。结果显示,目标化合物多潘立酮、莫沙必利和西沙必利在线性范围内线性关系良好,相关系数(r)≥0.9991,检出限为0.08 μg/kg,定量限为0.20 μg/kg,实际空白样品加标回收率在80.5%~110.5%之间,变异系数为0.83%~7.55%。结果表明,该方法检测效率高、检测结果准确,适用于动物源性食品中3种胃动力药物的快速检测。
关键词 超高效液相色谱-三重四极杆-在线离子阱质谱;动物源性食品;胃动力药物;检测
Determination of Three Supplementary Chemical Drugs in Food Products of Animal Origin by UPLC- MS/MS -QTRAP
ZHANG Xian-Chen1 HU Yi-Guang1 QI He-Ming2 LI Yong1 LIANG Na-Na2
Abstact This study established an ultra-high performance liquid chromatography tandem-linear ion trap mass spectrometry (UPLC-MS/MS-QTRAP) method for the determination of three supplementary chemical drugs (domperidone, mosapride and cisapride) in food products of animal origin, including meat from livestock, fish, and animal organs. The samples were extracted with ethyl acetate, concentrated and re-dissolved. The separation of the three target compounds was performed on an Agilent poroshell C18 column (100 mm×3.0 mm, 2.7 μm), using a gradient elution of methanol and 20 mmol/L amine acetate solution as the mobile phase. The target compounds were detected in positive electrospray ionization (ESI) using multiple reaction monitoring (MRM) mode, and quantified using the internal standard method. The result showed that the calibration curves of the three supplementary chemical drugs showed good linearity (r≥0.9991). The detection limits were 0.08 μg/kg, and the quantitation limits were 0.20 μg/kg. The recoveries of actual blank spiked sample ranged from 80.5% to 110.5%, with a coefficient of variation (CV) between 0.83% and 7.55%. This method has higher analysis efficiency, higher sensitivity and accuracy, making it suitable for the rapid detection of these three supplementary chemical drugs in food products of animal origin.
Keywords ultra-high performance liquid chromatography-tandem/linear ion trap mass spectrometry (UPLC-MS/MS-QTRAP); food products of animal origin;supplementary chemical drugs; determination
基金项目:拱北海关科技项目(2023GK016)
第一作者:张宪臣(1976—),男,汉族,吉林大安人,硕士,正高级工程师,主要从事食品安全与质量控制工作,E-mail: 496758046@qq.com
1. 中山海关技术中心 中山 528403
2. 中国海关科学技术研究中心 北京 100026
1. Technology Center of Zhongshan Customs, Zhongshan 528403
2. Science and Technology Research Center of China Customs, Beijing 100026
在兽医领域中,多潘立酮、莫沙必利和西沙必利曾是集约化养殖过程中常用的增加胃动力的药物。其中,多潘立酮商品名又称吗丁啉[1];莫沙必利又称美唯宁,是一种碱,与酸反应成盐后更加稳定,而所用酸以枸橼酸居多,故莫沙必利也叫枸橼酸莫沙必利(Mosapride Citrate)[3]。多潘立酮、莫沙必利和西沙必利能够刺激胃肠道平滑肌收缩,促进胃肠道蠕动,加速食物在胃肠道内排空[2-3],多用于治疗消化不良引起的各种消化性胃肠道疾病。3种药物主要用于医治牛、羊、猪等大型牲畜的急性胃肠鼓气、前胃弛缓等胃肠疾病,多潘立酮还可以对鱼类进行催产,具有诱导养殖鱼类排卵和产卵的功效。虽然多潘立酮、莫沙必利和西沙必利在胃肠疾病方面有较好的治疗效果,但是其使用不当也会产生一些副作用。多潘立酮由于可能会引发心律失常、心绞痛、心肌梗死等心脏疾病,欧美等地区的多个国家已禁止该药物的使用[4]。西沙必利由于具有心脏毒性,会造成患者严重的心律失常因而退出市场,现临床中严禁使用该药物[5-6]。莫沙必利可能导致服用者肝功能异常、损害中枢神经系统。2022年5月31日,日本厚生劳动省发布生食发0531第1号公告,修订了《食品和食品添加剂标准》,其中新增牛肉、牛脂肪中莫沙必利的限量要求(限量为0.01 mg/kg),修改其他陆生哺乳动物肉中莫沙必利的限量要求(限量由0.02 mg/kg修改为0.04 mg/kg)[7]。
目前胃动力类药物多潘立酮、莫沙必利和西沙必利的检测对象主要集中在口服药、中药、保健品和血浆中[8-14],经知网查询,国内未检索到动物源性食品中这三种药物的检测方法和相关标准,因此,建立灵敏可靠的检测动物源性食品中多潘立酮、莫沙必利和西沙必利的分析方法具有重要意义。当前采用的前处理方法主要是超声提取,采用的仪器检测方法有容量分析法、分光光度法[15-16]、间接原子吸收光谱法[17]、薄层色谱法[18]、液相色谱法[19-21]、液质联用法[22-24]等。其中,容量分析法和分光光度法由于专属性不强,仅适用于单一组分的分析。薄层色谱法是一种半定量的检测方法,易受外界环境影响。间接原子吸收光谱法通过检测氯的含量对目标化合物进行间接检测,检出限较高,溶液、温度等因素对检测结果影响较大。HPLC虽然分离能力较强,但是无法对目标化合物进行有效的确证。LC-MS/MS样品前处理简单,检出限低,灵敏度高,能够较为准确地对目标化合物进行定量定性分析。
本文采用乙酸乙酯提取样品,利用超高效液相色谱-三重四极杆-线性离子阱质谱建立了畜肉、鱼肉及各种动物内脏等动物源性食品中胃动力类药物多潘立酮、莫沙必利和西沙必利的残留量检测方法。
1 实验部分
1.1 材料、试剂与仪器
材料:部分样品为中山市口岸监管部门送检的法检样品,部分样品由中山市超市和农贸市场购买获得。
标准品:多潘立酮(纯度≥99.5%)、莫沙必利(纯度≥99.9%)、西沙必利(纯度≥99.9%)购于天津ALTA科技有限公司,多潘立酮同位素内标(纯度≥96.0%)、莫沙必利同位素内标(纯度≥98.0%)、西沙必利同位素内标(纯度≥95.0%)购于加拿大Trc公司。
试剂:甲醇、乙腈、乙酸乙酯(色谱纯,美国Fisher公司);OasisPRiME HLB固相萃取小柱(规格:6 CC/200 mg,美国Waters公司);乙酸铵(色谱纯,上海安谱科技有限公司)。实验用超纯水为Milli-Q超纯水仪(美国Mimpore公司)制得。
仪器:AB5500+三重四极杆/线性离子阱质谱仪,配有Analyst工作站,Waters ARC超高效液相色谱仪,配有Empower工作站(美国Waters公司);Sigma 3K15台式冷冻高速离心机(美国Sigma公司);Syncore平行定量浓缩系统(含DLSB recirculatingchiller系统、V-700/701真空泵)(瑞士BUCHI公司)。
1.2 标准溶液配制
用甲醇分别配制多潘立酮、莫沙必利、西沙必利、多潘立酮同位素内标、莫沙必利同位素内标和西沙必利同位素内标标准品储备液,质量浓度为1.0 mg/mL。
用甲醇稀释适量的多潘立酮、莫沙必利和西沙必利储备液,配成各相应质量浓度为1.0 μg/mL的混合标准工作液。
用甲醇稀释适量的多潘立酮同位素内标、莫沙必利同位素内标和西沙必利同位素内标储备液,配成各相应质量浓度为0.1 μg/mL的混合标准工作液。
1.3 液相色谱和串联质谱条件
1.3.1 色谱条件
色谱柱:Agilent poroshell C18(3.0 mm×100 mm,2.7 μm);柱温箱设定温度:40℃;进样体积:10 μL;20 mmol/L乙酸铵水溶液设定为流动相A、甲醇设定为流动相B;流速为0.4 mL/min。梯度洗脱程序:初始设定流动相B比例35%,保持2.0 min;之后在2.0 min内,流动相B的比例由35%线性减少至8%;流动相B比例8%,保持3.0 min后,流动相B的比例在0.1 min内由8%增至35%,最后流动相B的比例35%,保持3.0 min。
1.3.2 质谱条件
离子源类型:电喷雾离子源;气帘气(氮气)流量:30.0 L/h;雾化器(氮气)流量:55.0 L/h;辅助气(Gas 2)流量:55.0 L/h;电喷雾电压:5500 V;离子源温度(TEM):550℃;扫描模式:正离子;检测方式:多反应监测(MRM)结合增强子离子扫描(EPI);目标化合物色谱、质谱等相关参数信息见表1。
1.4 样品预处理
取样品(包括畜肉、鱼肉、牛肚、羊肚等动物内脏)中具有代表性的可食部分约500 g,用组织捣碎机充分打碎,混匀,装入洁净的容器内,密封并做好标识,于-18℃或以下保存。准确称取制备好的样品2.00 g于50 mL具塞高速离心管中,准确加入50 μL混合内标工作液,涡旋振荡30 s,加入10 mL水,用均质器(10000 r/min)均质2 min,5 mL水清洗刀头,加入10 mL乙酸乙酯,涡旋振荡30 s,超声提取10 min后取出,4500 r/min离心10 min,取上清液于50 mL离心管中;剩余溶液中加入10 mL乙酸乙酯,重复提取1次,合并2次提取液于40℃平衡定量浓缩仪中浓缩至干。1.0 mL甲醇水溶液(4+6,v/v)复溶残渣,再加入3.0 mL正己烷,涡旋振荡30 s,最后取1.0 mL底层溶液在温度10℃、转速12000 r/min的条件下离心10 min,上清液过0.22 μm微孔有机滤膜后供液相色谱-质谱/质谱仪测定。
2 结果与分析
2.1 色谱条件优化
本文选择实验室通用性较好的4款色谱柱Waters AtiantisRT3(150 mm×2.1 mm,3.3 μm)、ThermoHypersil GOLD C18(100 mm×2.1 mm,1.9 μm)、RESTEK Ultra AQ C18 (100 mm×2.1mm,3.1 μm)和Agilent poroshell C18 (100 mm×3.0 mm,2.7 μm)对3种目标化合物进行分离,结果显示,RESTEK Ultra AQ C18(×2.1 mm,3.1 μm)和Waters AtiantisRT3(150 mm×2.1mm,3.3 μm)色谱柱虽然可以对3种目标化合物进行分离,但是3种目标化合物色谱峰较宽,且多潘立酮和莫沙必利色谱峰有轻微分裂;ThermoHypersil GOLDC18(100 mm×2.1mm,1.9 μm)色谱柱由于粒径较小,在流动相流速0.4 mL/min的条件下,系统压力超过9000 psi,调整流速为0.25 mL/min后,3种目标化合物色谱峰能够有效达到基线分离,但是多潘立酮色谱峰出现拖尾现象;Agilent poroshell C18(100 mm×3.0 mm,2.7 μm)由于其填料由1.7 μm直径的实心核和0.5 μm厚的多孔外层构成,3种目标化合物在Agilent poroshell C18 (100 mm×3.0 mm,2.7 μm)形成的色谱峰峰形尖锐,对称性好(图1)。
2.2 质谱条件优化
将3种胃动力药物及内标单一标准品溶液以7.0 μL/min的速度直接注入质谱仪,在正离子模式下进行全扫描(Scan)以确定母离子;二级扫描(SIM)模式下,确定定性离子和定量离子;与已确定的母离子组成多反应监测方法(MRM)离子对;对得到的离子对进行优化,确定最佳的碎裂电压(CE)、去簇电压(DP)(表1)。在实际样品检测中,由于MRM扫描模式在目标化合物确证中有一定的缺陷,特别是对于基质较复杂动物源性样品容易出现误判,本研究采用在线离子阱质谱对阳性样品进行二次确证,即采用MRM-IDA-EPI扫描模式,MRM扫描模式用于目标化合物的定量,EPI(增强子离子扫描)模式得到的高质量目标化所有特征性碎片,用于目标化合物的二次定性。QTRAP在EPI扫描模式下可以兼顾低碰撞能量、中等碰撞能量和高碰撞能量的子离子或者化合物(图2)。
2.3 前处理条件优化
2.3.1 提取溶剂的优化
选取甲醇、乙腈、80%乙腈饱和氯化钠水溶液、乙酸乙酯等4种试剂作为提取剂,结果显示:用乙醇和甲醇溶液对样品中3种目标化合物进行直接提取时,3种目标化合物的回收率低于40%,未达到检测要求;用80%乙腈饱和氯化钠水溶液提取时,多潘立酮的回收率低于80%,未达到检测要求。本研究考虑到乙酸乙酯易于与水分层,首先加入10 mL水,用均质器(10000 r/min)对样品均质处理,再加入乙酸乙酯进行超声提取,离心后对上清液进行浓缩处理,最终结果显示,3种目标化合物回收率在80%~110%之间,因此研究选择乙酸乙酯作为提取溶液(图3)。
2.3.2 固相萃取柱的选择
被测样品中含有蛋白质、脂肪、磷脂、碳水化合物、矿物质、维生素等成分,因此本研究选择Oasis HLB固相萃取小柱、Oasis PRiME HLB固相萃取小柱、Oasis MCX固相萃取交换柱和不过萃取小柱4种方式对前处理净化效果进行考察,通过阴性样品添加混合标准品溶液的方式对4种方式的净化效果进行比较分析,添加浓度为0.2 μg/kg,每个样品3平行,外标法定量,阳性样品峰面积与相同浓度的标准品峰面积进行比值,结果用于评价净化效果。结果显示,Oasis MCX固相萃取交换柱虽然能够去除99%的磷脂,但是对其他干扰物质的净化效果有限,目标化合物中多潘立酮在洗脱过程中损失较大,主要是由于多潘立酮化学结构比较独特,是由苯基环和吗啡环组成,对Oasis MCX固相萃取交换柱吸附造成一定影响。Oasis PRiME HLB固相萃取柱和Oasis HLB固相萃取柱作用机理相似,都是反相固相萃取柱,其中Oasis PRiME HLB固相萃取柱能够去除95%的干扰物,实验结果显示,Oasis PRiME HLB固相萃取柱的净化效果要高于Oasis HLB固相萃取柱,但是在净化过程中多潘立酮的损失超过50%。本研究尝试不过萃取小柱,采用乙酸乙酯对样品进行液液萃取,浓缩复溶后,采用添加正己烷和高速离心的方式去除干扰物质,3种目标化合物的损失小于30%。综合比较4种前处理方法,考虑到实际净化效果和检测成本的因素,本文选择不过固相萃取柱的方式对畜肉、鱼肉及动物内脏样品进行净化处理(图4)。
图4 不同净化方式对3种胃动力药物残留的影响
Fig.4 Effect of different extraction methods on the residue of the three supplementary chemical drugs
2.3.3 浓缩条件的优化和离心条件
由于3种胃动力药物易受温度、压力等因素影响,因此本研究在减压的条件下采用平行样品定量浓缩仪对过Oasis PRiME HLB固相萃取柱的样品提取液进行浓缩处理,对浓缩过程中各项参数进行了优化,确定的最终浓缩条件是浓缩温度50℃;冷凝温度0℃;真空下降梯度为保持250 kPa真空度2 min,真空度下降至80 kPa后保持5 min,真空度下降至20 kPa后浓缩至干。使用平行样品定量浓缩仪进行浓缩,能够有效地降低检测成本,提高浓缩效率和质量,该设备能够同时浓缩处理24份样品提取液,仅需0.5 h即可完全浓缩至干。
2.4 基质效应
畜肉、畜肉内脏、鱼肉等动物源性食品中含有脂肪、蛋白质、矿物质和维生素等成分,可能存在基质抑制或基质增强效应,因此本研究对其进行了基质效应(Matrix Effect,ME)评价。采用基质标准曲线斜率与空白溶剂标准曲线斜率的相对大小来考察基质效应,计算公式为ME =(K基质/K溶剂-1)×100%。选取不含有3种目标化合物的空白样品进行基质效应的考察,按1.4方法操作,得到空白基质液,再分别以甲醇和空白基质液配制标准系列溶液,进样测定,绘制标准曲线,得到曲线斜率计算基质效应,如果ME值为正,表明基质对检测结果具有增强效应,如果为负,表明基质对检测结果具有抑制效应。检测结果显示3种目标化合物的ME值在2.9%~12%之间(图5),说明畜肉、畜肉内脏、鱼肉样品对3种目标化合物存在轻微的基质抑制效应,为了保证本方法的严谨性,研究采用内标法对畜肉、畜肉内脏、鱼肉等动物源性食品中3种胃动力药物进行定量分析。
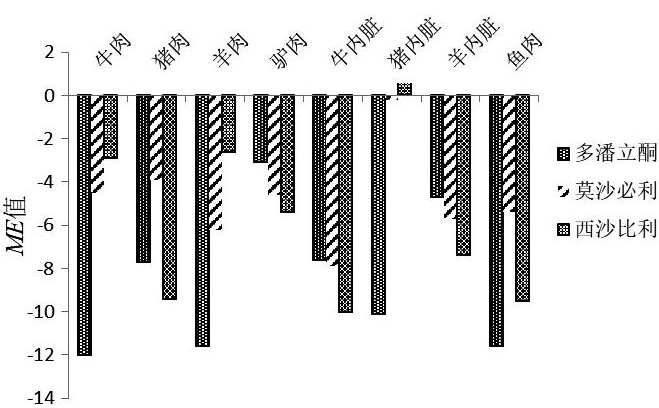
图5 3种目标化合物ME值比较
Fig.5 Comparison of ME value of the three drug
2.5 方法学验证
2.5.1 标准曲线、线性范围和检出限
将甲醇稀释混合标准工作液分别配制成浓度为0.05 μg/L、0.1 μg/L、0.5 μg/L、1.0 μg/L、5.0 μg/L的标准工作液,浓度由低到高进样检测,并根据检测结果绘制标准工作曲线。结果显示3种目标化合物在0.05~5.0 μg/L浓度范围内线性良好,相关系数均大于0.9991(表2)。
按照1.3和1.4进行加标实验并重复6次,分别获得6次进样的信噪比,以10倍信噪比对应目标物的质量浓度为方法定量限(LOQ),3倍信噪比对应目标物的质量浓度为方法检出限(LOD),确定3种目标化合物的检出限为0.08 μg/kg,定量限为0.2 μg/kg(表2)。
2.5.2 方法准确度和精密度
依据GB/T 27404—2008《实验室质量控制规范 食品理化检测》对方法准确度和精密度的实验方案进行设计,在方法定量限、2倍定量限和10倍定量限进行3水平试验,即本研究对阴性动物源性食品中添加混合标准品溶液的浓度水平分别为0.2 μg/kg,0.4 μg/kg,2.0 μg/kg,每个水平6平行。结果显示,3种目标化合物的回收率在80.5%~110.5%之间,满足被测组分浓度<0.1 mg/kg(即100 μg/kg)时,回收率应在60%~120%的技术要求。对每个水平6平行加标样品的试验数据进行统计,计算变异系数,结果显示,3种目标化合物的变异系数在0.83%~7.55%之间,符合满足被测组分浓度为10 μg/kg时,变异系数应<21%的技术要求(表3)。
3 结论
本研究以乙酸乙酯溶液为萃取剂对样品进行提取,浓缩后经正己烷和高速冷冻离心去除提取液中的干扰物质,建立了动物源性食品中3种胃动力药物的超高效液相色谱-三重四极杆-在线离子阱质谱的快速检测方法,方法灵敏度高、操作简单、适用性强,适用于相关实验室对大批量动物源性食品样品进行检测。
参考文献
[1]吴诗聪. 多潘立酮的药理、药效及临床应用[J]. 临床合理用药杂志. 2012. 5(7):86-87.
[2] Katayama K. Morio Y. Haga K. et al. Cisapride, a gastroprokinetic agent, binds to 5-HT4 receptors[J]. Nihon Yakurigaku Zasshi. Folia pharmacologica Japonica. 1995, 105(6): 461-468.
[3]沈雷鸣. 西沙必利对失血性休克下肠道屏障功能保护作用的实验研究及机制探讨[D]. 吉林: 吉林大学, 2016.
[4]陈静. 雷尼替丁联合多潘立酮治疗小儿消化不良的临床分析[J]. 中国医药指南, 2022, 20(30): 65-67.
[5] Layton D. Key C. Shakir S. Prolongation of the QT interval and cardiac arrhythmias associated with cisapride:Limitations of the pharmacoepidemiological studies conducted and proposals for the future[M]. Pharmacoepidemiology and Drug Safety, 2003, 12(1): 31-40.
[6] Al-Judaibi B. Chande N. Gregor J. Safety and efficacy of tegaserod therapy in patients with irritable bowel syndrome or chronic constipation[J]. Can J ClinPharmacol. 2010. 17(1): 194-200.
[7]日本厚生劳动省:修订《食品和食品添加剂标准》(生食发0531第1号公告)[EB/OL]. https://www.mhlw.go.jp/content/001102261.pdf
[8]赵超群, 李樱红, 罗金文. 超高效液相色谱-串联质谱测定保健食品中添加的10种化学药物[J]. 药物分析杂志, 2014, 34(9): 1627-1633.
[9]李锐, 张海防. UPLC-MS/MS检测中药及保健品中添加的9种改善胃肠功能类化学药物[J]. 中医药导报, 2018, 24(10): 96-99.
[10] 陈红. 高效液相梯度洗脱法测定西沙必利的含量[J]. 浙江中医药大学学报, 2002, 26(2): 70-71.
[11] 刘剑, 何志春, 荆强. HPLC法测定枸橼酸莫沙比利片的含量[J]. 中国药师, 2006, 9(2): 143-144.
[12] 单爱莲, 马静, 李浩. 液相色谱-串联质谱法测定人血浆中多潘立酮的浓度[J]. 中国临床药理学杂志, 2018, 34(18): 2211-2213.
[13]谢君, 吕玲燕, 冯星月, 等. UPLC-MS/MS法测定多潘立酮口腔崩解片的含量[J]. 实用药物与临床, 2018, 21(3): 302-304.
[14] 白娜. 利用快检车近红外光谱鉴别多潘立酮片[J]. 现代盐化工, 2023, 50(4): 67-68.
[15] 郭琦. 张兰芬. 陈慧娟. 等. 西沙必利片的荧光分光光度法测定[J]. 中国药师, 2007, 1: 54-55.
[16]马宁,谢安云, 何燕. 紫外分光光度法测定西沙必利片的含量[J].中国药师, 2000, 6: 59-60.
[17]冒爱荣. 间接原子吸收光谱法测定西沙必利片的含量[J]. 应用化学, 2009, 26(2): 246-248.
[18]余红, 张继红, 金贤武. 抗溃疡类中成药非法添加化学物质薄层色谱检测方法研究[J]. 江西中医药大学学报, 2012, 24(5): 65-66.
[19]黄淑英, 张敏, 陈可师. HPLC法测定西沙必利片的含量[J]. 化学分析计量, 2003, 12(5): 24-25.
[20]贺玉峰. HPLC法测定多潘立酮片的含量[J]. 中国药品标准, 2006, 7(4): 41-42.
[21]陆麟, 吴佳伟, 华璇洁, 等. HPLC法测定枸橼酸莫沙必利的稳定性[J]. 临床合理用药杂志, 2015, 8(8A): 124-125.
[22] 潘群, 秦永平, 向瑾, 等. HPLC-MS/MS法测定人血浆中莫沙必利及其代谢物含量[J]. 药物分析杂志, 2009. 29(11): 1842-1845.
[23]郝光涛, 董瑞华, 高洪志, 等. HPLC-MS-MS法定量测定人血浆中多潘立酮的浓度[J]. 中国药物应用与监测, 2008, 5(5): 19-21.
[24]王冬, 卢丹, 张兴哲. 超高效液相色谱-质谱联用法测定多潘立酮的含量[J]. 中国药物评价, 2019, 36(2): 97-100.
表1 多潘立酮、莫沙必利和西沙必利及同位素内标化合物仪器参数
Table 1 Parameters of instrument for Dompridone, Mosapride, Cisapride, Dompridone-d6, Mosapride-d5 and Cisapride-13C,d3
目标物 | (min) | CAS号 | 化学式 | 分子量 | (m/z) | (m/z) | (eV) | (eV) |
多潘立酮 | 3.07 | 57808-66-9 | C22H24CIN5O2 | 425.91 | 426.1 | 175.2* | 29 | 36 |
147.1 | 29 | 46 | ||||||
莫沙必利 | 4.40 | 112885-41-3 | C21H25CIFN3O3 | 421.89 | 422.1 | 198.1* | 26 | 28 |
170.2 | 26 | 51 | ||||||
西沙必利 | 3.93 | 81098-60-4 | C23H29CIFN3O4 | 465.95 | 466.1 | 184.2* | 17 | 34 |
234.2 | 17 | 32 | ||||||
多潘立酮-d6 | 2.93 | 1329614-18-7 | C22H18D6CIN5O2 | 431.95 | 432.2 | 181.2* | 61 | 39 |
莫沙必利-d5 | 4.34 | 1246820-66-5 | C21H20D5CIFN3O3 | 426.93 | 427.2 | 203.2* | 32 | 26 |
西沙必利-13C,d3 | 3.90 | 1285970-69-5 | C2213CH26D3CIFN3O4 | 469.96 | 470.2 | 188.0* | 30 | 42 |
注: 带“*”的离子为定量离子
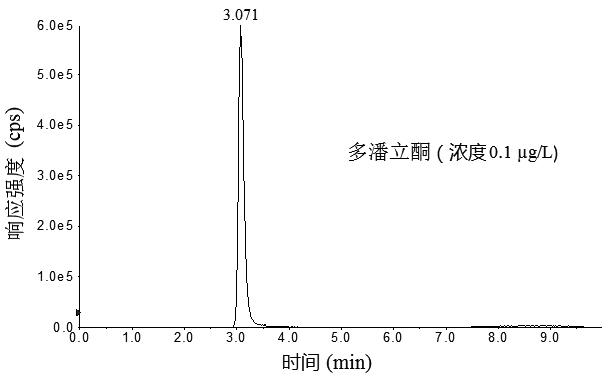
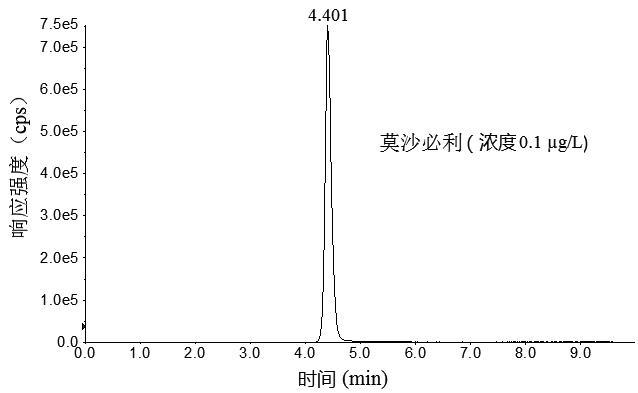
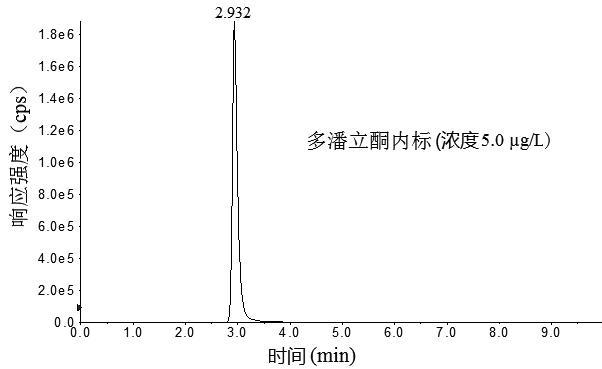
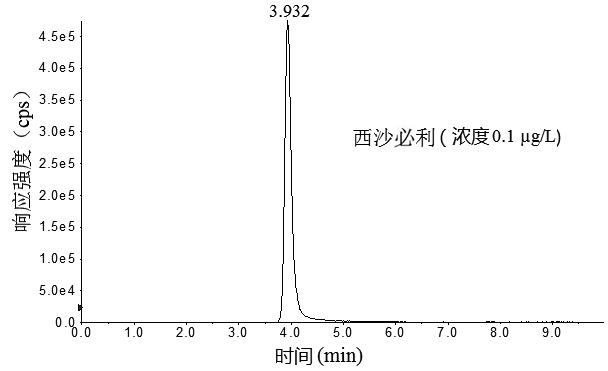
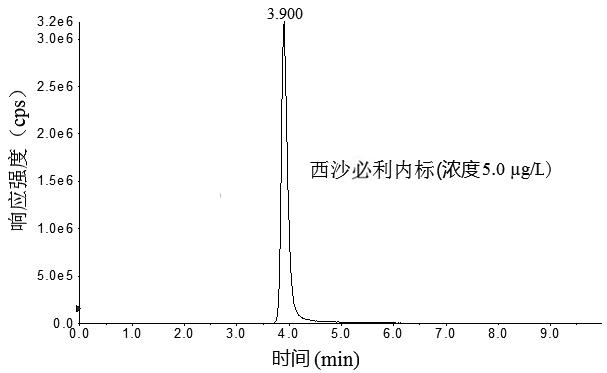
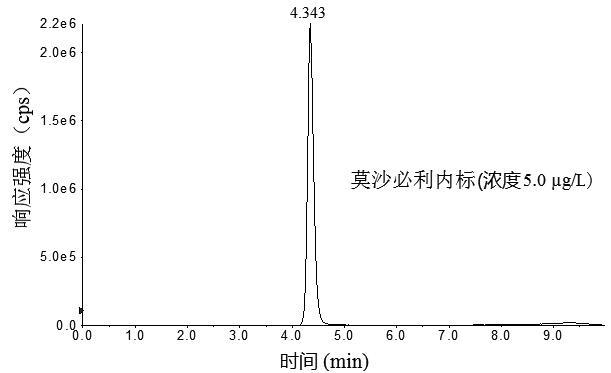
图1 3种胃动力药物及内标物混合标准溶液色谱图
Fig.1 Chromatograms of mixed standard solution of the three supplementary chemical drugs and internal standards
图2 3种目标化合物EPI扫描质谱图
Fig.2 Mass spectrum of the three drug obtained by EPI

表2 3种目标物线性回归方程和相关系数
Table 2 Linear regression equations and correlation coefficients for the three compounds
目标化合物 | 线性范围 (μg/L) | 线性方程 | 相关系数 r | 检出限 (μg/k g ) | 定量限 (μg/k g ) |
多潘立酮 | 0.05~5.0 | Y = 0. 157 X + 0.00 107 | 0.999 5 | 0.08 | 0.2 |
莫沙必利 | 0.05~5.0 | Y = 0. 155 X + 0.00 151 | 0.999 1 | 0.08 | 0.2 |
西沙必利 | 0.05~5.0 | Y = 0.0809 X + 0.000243 | 0.999 9 | 0.08 | 0.2 |
注: X为目标化合物峰面积; Y为目标化合物浓度 (μg /L).
表3 3种药物残留加标回收率和变异系数(n = 6)
Table 3 Spiked recoveries and CV of the three compounds in blank samples (n = 6)
基质 | 化合物 | (μg/kg) | (%) | (%) | 基质 | 化合物 | (μg/kg) | (%) | (%) |
牛肉 | 多潘立酮 | 0.2, 0.4, 2.0 | 80.5~93.5 | 0.83~4.82 | 鱼肉 | 多潘立酮 | 0.2, 0.4, 2.0 | 92.8~104.1 | 1.33~3.62 |
莫沙必利 | 0.2, 0.4, 2.0 | 92.5~100.5 | 1.21~3.19 | 莫沙必利 | 0.2, 0.4, 2.0 | 86.3~101.0 | 0.85~2.78 | ||
西沙必利 | 0.2, 0.4, 2.0 | 88.5~102.5 | 1.67~4.77 | 西沙必利 | 0.2, 0.4, 2.0 | 84.3~100.9 | 1.97~3.38 | ||
猪肉 | 多潘立酮 | 0.2, 0.4, 2.0 | 80.5~91.3 | 1.42~4.11 | 牛肠 | 多潘立酮 | 0.2, 0.4, 2.0 | 97.6~107.0 | 1.31~2.27 |
莫沙必利 | 0.2, 0.4, 2.0 | 84.0~98.0 | 2.37~3.17 | 莫沙必利 | 0.2, 0.4, 2.0 | 90.8~107. 5 | 1.31~2.27 | ||
西沙必利 | 0.2, 0.4, 2.0 | 87.5~103.3 | 1.95~4.05 | 西沙必利 | 0.2, 0.4, 2.0 | 95.0~105.5 | 1.42~2.62 | ||
羊肉 | 多潘立酮 | 0.2, 0.4, 2.0 | 86.8~102.5 | 1.33~4.33 | 牛肚 | 多潘立酮 | 0.2, 0.4, 2.0 | 82.3~102.5 | 2.56~4.68 |
莫沙必利 | 0.2, 0.4, 2.0 | 90.5~109.8 | 1.56~7.55 | 莫沙必利 | 0.2, 0.4, 2.0 | 97.5~108.5 | 2.77~3.71 | ||
西沙必利 | 0.2, 0.4, 2.0 | 81.3~107.5 | 2.10~7.06 | 西沙必利 | 0.2, 0.4, 2.0 | 96.0~110.5 | 1.50~4.26 | ||
驴肉 | 多潘立酮 | 0.2, 0.4, 2.0 | 92.8~107.3 | 2.20~2.79 | 羊肚 | 多潘立酮 | 0.2, 0.4, 2.0 | 90.8~102.0 | 2.01~5.19 |
莫沙必利 | 0.2, 0.4, 2.0 | 84.8~109.0 | 1.49~7.55 | 莫沙必利 | 0.2, 0.4, 2.0 | 84.6~100.0 | 3.69~5.22 | ||
西沙必利 | 0.2, 0.4, 2.0 | 83.0~107.5 | 2.47~7.06 | 西沙必利 | 0.2, 0.4, 2.0 | 83.1~95.0 | 2.31~5.44 |
